Paid employment among women is growing worldwide. For example, almost 70% of women in the United States are employed outside the home during their predominant childbearing years (ages 20 to 34). Furthermore, since the 1940s there has been an almost linear trend in synthetic organic chemical production, creating a more hazardous environment for the pregnant worker and her offspring.
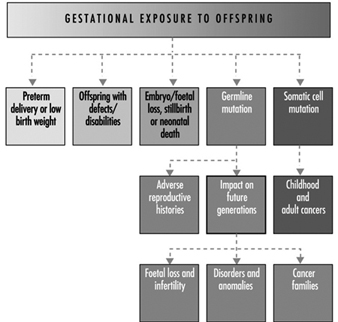
Ultimately, a couple’s reproductive success depends on a delicate physiochemical balance within and between the father, the mother and the foetus. Metabolic changes occurring during a pregnancy can increase exposure to hazardous toxicants for both worker and concetus. These metabolic changes include increased pulmonary absorption, increased cardiac output, delayed gastric emptying, increased intestinal motility and increased body fat. As shown in figure 1, exposure of the concetus can produce varying effects depending on the phase of development—early or late embryogenesis or the foetal period.
Figure 1. Consequences of maternal exposure to toxicants on the offspring.
Transport time of a fertilized ovum before implantation is between two and six days. During this early stage the embryo may be exposed to chemical compounds that penetrate into the uterine fluids. Absorption of xenophobic compounds may be accompanied by degenerative changes, alteration in the blastocystic protein profile or failure to implant. Insult during this period is likely to lead to a spontaneous abortion. Based on experimental data, it is thought that the embryo is fairly resistant to teratogenic insult at this early stage because the cells have not initiated the complex sequence of chemical differentiation.
The period of later embryogenesis is characterized by differentiation, mobilization and organization of cells and tissue into organ rudiments. Early pathogenesis may induce cell death, failed cellular interaction, reduced biosynthesis, impaired morphogenic movement, mechanical disruption, adhesions or oedema (Paul 1993). The mediating factors that determine susceptibility include route and level of exposure, pattern of exposure and foetal and maternal genotype. Extrinsic factors such as nutritional deficiencies, or the additive, synergistic or antagonistic effects associated with multiple exposures may further impact the response. Untoward responses during late embryogenesis may culminate in spontaneous abortion, gross structural defects, foetal loss, growth retardation or developmental abnormalities.
The foetal period extends from embryogenesis to birth and is defined as beginning at 54 to 60 gestational days, with the concetus having a crown-rum length of 33 mm. The distinction between the embryonic and foetal period is somewhat arbitrary. The foetal period is characterized developmentally by growth, histogenesis and functional maturation. Toxicity may be manifested by a reduction in cell size and number. The brain is still sensitive to injury; myelination is incomplete until after birth. Growth retardation, functional defects, disruption in the pregnancy, behavioural effects, translacental carcinogenesis or death may result from toxicity during the foetal period. This article discusses the biological, sociological and epidemiological effects of maternal environmental/occupational exposures.
Embryonic/Foetal Loss
The developmental stages of the zygote, defined in days from ovulation (DOV), proceed from the blastocyst stage at days 15 to 20 (one to six DOV), with implantation occurring on day 20 or 21 (six or seven DOV), to the embryonic period from days 21 to 62 (seven to 48 DOV), and the foetal period from day 63 (49+ DOV) until the designated period of viability, ranging from 140 to 195 days. Estimates of the probability of pregnancy termination at one of these stages depend on both the definition of foetal loss and the method used to measure the event. Considerable variability in the definition of early versus late foetal loss exists, ranging from the end of week 20 to week 28. The definitions of foetal and infant death recommended by the World Health Organization (1977) are listed in table 1. In the United States the gestational age setting the lower limit for stillbirths is now widely accepted to be 20 weeks.
Table 1. Definition of foetal loss and infant death
| Spontaneous abortion | ≤500 g or 20-22 weeks or 25 cm length |
| Stillbirth | 500 g (1000 g International) nonviable |
| Early neonatal death | Death of a live-born infant ≤7 days (168 hours) |
| Late neonatal death | 7 days to ≤28 days |
Source: World Health Organization 1977.
Because the majority of early aborted foetuses have chromosomal anomalies, it has been suggested that for research purposes a finer distinction should be made—between early foetal loss, before 12 weeks’ gestation, and later foetal loss (Källén 1988). In examining late foetal losses it also may be appropriate to include early neonatal deaths, as the cause may be similar. WHO defines early neonatal death as the death of an infant aged seven days or younger and late neonatal death as occurring between seven and 29 days. In studies conducted in developing countries, it is important to distinguish between prepartum and intrapartum deaths. Because of problematic deliveries, intrapartum deaths account for a large portion of stillbirths in less developed countries.
In a review by Kline, Stein and Susser (1989) of nine retrospective or cross-sectional studies, the foetal loss rates before 20 weeks’ gestation ranged from 5.5 to 12.6%. When the definition was expanded to include losses u to 28 weeks’ gestation, the foetal loss rate varied between 6.2 and 19.6%. The rates of foetal loss among clinically recognized pregnancies in four prospective studies, however, had a relatively narrow range of 11.7 to 14.6% for the gestational period u to 28 weeks. This lower rate, seen in prospective versus retrospective or cross-sectional designs, may be attributable to differences in underlying definitions, misreporting of induced abortions as spontaneous or misclassification of delayed or heavy menses as foetal loss.
When occult abortions or early “chemical” losses identified by an elevated level of human chorionic gonadotrohins (hCG) are included, the total spontaneous abortion rate jumps dramatically. In a study using hCG methods, the incidence of post-implantation subclinical loss of fertilized ova was 22% (Wilcox et al. 1988). In these studies urinary hCG was measured with immunoradiometric assay using a detection antibody. The assay originally used by Wilcox employed a now extinct high affinity, polyclonal rabbit antibody. More recent studies have used an inexhaustible monoclonal antibody that requires less than 5 ml of urine for replicate samples. The limiting factor for use of these assays in occupational field studies is not only the cost and resources needed to coordinate collection, storage and analysis of urine samples but the large population needed. In a study of early pregnancy loss in women workers exposed to video display terminals (VDTs), approximately 7,000 women were screened in order to acquire a usable population of 700 women. This need for ten times the population size in order to achieve an adequate sample stems from reduction in the available number of women because of ineligibility due to age, sterility and the enrollment exclusively of women who are using either no contraceptives or relatively ineffective forms of contraception.
More conventional occupational studies have used recorded or questionnaire data to identify spontaneous abortions. Recorded data sources include vital statistics and hospital, private practitioner and outpatient clinic records. Use of record systems identifies only a subset of all foetal losses, principally those that occur after the start of prenatal care, typically after two to three missed periods. Questionnaire data are collected by mail or in personal or telephone interviews. By interviewing women to obtain reproductive histories, more complete documentation of all recognized losses is possible. Questions that are usually included in reproductive histories include all pregnancy outcomes; prenatal care; family history of adverse pregnancy outcomes; marital history; nutritional status; re-pregnancy weight; height; weight gain; use of cigarettes, alcohol and prescription and nonprescription drugs; health status of the mother during and prior to a pregnancy; and exposures at home and in the workplace to physical and chemical agents such as vibration, radiation, metals, solvents and pesticides. Interview data on spontaneous abortions can be a valid source of information, particularly if the analysis includes those of eight weeks’ gestation or later and those that occurred within the last 10 years.
The principal physical, genetic, social and environmental factors associated with spontaneous abortion are summarized in table 2. To ensure that the observed exposure-effect relationship is not due to a confounding relationship with another risk factor, it is important to identify the risk factors that may be associated with the outcome of interest. Conditions associated with foetal loss include syphilis, rubella, genital Mycolasma infections, herpes simplex, uterine infections and general hyperpyrexia. One of the most important risk factors for clinically recognized spontaneous abortion is a history of pregnancy ending in foetal loss. Higher gravidity is associated with increased risk, but this may not be independent of a history of spontaneous abortion. There are conflicting interpretations of gravidity as a risk factor because of its association with maternal age, reproductive history and heterogeneity of women at different gravidity ranks. Rates of spontaneous abortion are higher for women younger than 16 and older than 36 years. After adjusting for gravidity and a history of pregnancy loss, women older than 40 were shown to have twice the risk of foetal loss of younger women. The increased risk for older women has been associated with an increase in chromosomal anomalies, particularly trisomy. possiblemale-mediated effects associated with foetal loss have been recently reviewed (Savitz, Sonnerfeld and Olshaw 1994). A stronger relationship was shown with paternal exposure to mercury and anaesthetic gases, as well as a suggestive but inconsistent relationship with exposure to lead, rubber manufacturing, selected solvents and some pesticides.
Table 2. Factors associated with small for gestational age and foetal loss
| Small for gestational age | |
| Physical-genetic | Environmental-social |
| Preterm delivery Multiple births Malformed foetus Hypertension Placental or cord anomaly Maternal medical history History of adverse pregnancy outcomes Race Chromosome anomalies Sex Maternal height, weight, weight gain Paternal height Parity Length of gestation Short interval between pregnancies |
Malnutrition Low income/poor education Maternal smoking Maternal alcohol consumption Occupational exposure Psychosocial stress Altitude History of infections Marijuana use |
| Foetal loss | |
| Physical-genetic | Environmental-social |
| Higher gravidity Maternal age Birth order Race Repeat spontaneous abortion Insulin dependent diabetes Uterine disorders Twinning Immunological factor Hormonal factors |
Socio-economic status Smoking history Prescribed and recreational drugs Alcohol use Poor nutrition Infections/maternal fever Spermicides Employment factors Chemical exposure Irradiation |
Employment status may be a risk factor regardless of a specific physical or chemical hazard and may act as a confounder in assessment of occupational exposure and spontaneous abortion. Some investigators suggest that women who stay in the workforce are more likely to have an adverse pregnancy history and as a result are able to continue working; others believe this group is an inherently more fit subpopulation due to higher incomes and better prenatal care.
Congenital Anomalies
During the first 60 days after conception, the developing infant may be more sensitive to xenobiotic toxicants than at any other stage in the life cycle. Historically, terata and congenital malformations referred to structural defects resent at birth that may be gross or microscopic, internal or external, hereditary or nonhereditary, single or multiple. Congenital anomaly, however, is more broadly defined as including abnormal behaviour, function and biochemistry. Malformations may be single or multiple; chromosomal defects generally produce multiple defects, whereas single-gene changes or exposure to environmental agents may cause either single defects or a syndrome.
The incidence of malformations depends on the status of the concetus—live birth, spontaneous abortus, stillbirth. Overall, the abnormality rate in spontaneous abortuses is approximately 19%, a tenfold increase in what is seen in the live born (Sheard, Fantel and Fitsimmons 1989). A 32% rate of anomalies was found among stillborn foetuses weighing more than 500 g. The incidence of major defects in live births is about 2.24% (Nelson and Holmes 1989). The prevalence of minor defects ranges between 3 and 15% (averaging about 10%). Birth anomalies are associated with genetic factors (10.1%), multifactorial inheritance (23%), uterine factors (2.5%), twinning (0.4%) or teratogens (3.2%). The causes of the remaining defects are unknown. Malformation rates are approximately 41% higher for boys than for girls and this is explained by the significantly higher rate of anomalies for male genital organs.
One challenge in studying malformations is deciding how to group defects for analysis. Anomalies can be classified by several parameters, including seriousness (major, minor), pathogenesis (deformation, disruption), associated versus isolated, anatomic by organ system, and aetiological (e.g., chromosomal, single gene defects or teratogen induced). Often, all malformations are combined or the combination is based either on major or minor categorization. A major malformation can be defined as one that results in death, requires surgery or medical treatment or constitutes a substantial physical or psychological handicap. The rationale for combining anomalies into large groups is that the majority arise, at approximately the same time period, during organogenesis. Thus, by maintaining larger sample sizes, the total number of cases is increased with a concomitant increase in the statistical power. If, however, the exposure effect is specific to a particular type of malformation (e.g., central nervous system), such grouping may mask the effect. Alternatively, malformations may be grouped by organ system. Though this method may be an improvement, certain defects may dominate the class, such as varus deformities of the feet in the musculoskeletal system. Given a sufficiently large sample, the optimal approach is to divide the defects into embryologically or pathogenetically homogenous groups (Källén 1988). Considerations should be given to the exclusion or inclusion of certain malformations, such as those that are likely caused by chromosomal defects, autosomal dominant conditions or malposition in utero. Ultimately, in analysing congenital anomalies, a balance has to be maintained between maintaining precision and compromising statistical power.
A number of environmental and occupational toxicants have been associated with congenital anomalies in offspring. One of the strongest associations is maternal consumption of food contaminated with methylmercury causing morphological, central nervous system and neurobehavioural abnormalities. In Japan, the cluster of cases was linked to consumption of fish and shellfish contaminated with mercury derived from the effluent of a chemical factory. The most severely affected offspring developed cerebral palsy. Maternal ingestion of polychlorinated biphenyl’s (CBs) from contaminated rice oil gave rise to babies with several disorders, including growth retardation, dark brown skin pigmentation, early eruption of teeth, gingival hyperplasia, wide sagittal suture, facial oedema and exophthalmoses. Occupations involving exposures to mixtures have been linked with a variety of adverse outcomes. The offspring of women working in the ul and aer industry, in either laboratory work or jobs involving “conversions” or aer refinement, also had increased risk of central nervous system, heart and oral cleft defects. Women working in industrial or construction work with unspecified exposures had a 50% increase in central nervous system defects, and women working in transportation and communication had two times the risk of having a child with an oral cleft. Veterinarians represent a unique group of health care personnel exposed to anaesthetic gases, radiation, trauma from animal kicks, insecticides and zoonotic diseases. Though no difference was found in the rate of spontaneous abortions or in birth weight of the offspring between female veterinarians and female lawyers, there was a significant excess of birth defects among veterinarians (Schenker et al. 1990). Lists of known, possible and unlikely teratogens are available as well as computer databases and risk lines for obtaining current information on potential teratogens (Paul 1993). Evaluating congenital anomalies in an occupational cohort is particularly difficult, however, because of the large sample size needed for statistical power and our limited ability to identify specific exposures occurring during a narrow window of time, primarily the first 55 days of gestation.
Small for Gestational Age
Among the many factors linked with infant survival, physical underdevelopment associated with low birth weight (LBW) resents one of the greatest risks. Significant weight gain of the foetus does not begin until the second trimester. The concetus weighs 1 g at eight weeks, 14 g at 12 weeks, and reaches 1.1 kg at 28 weeks. An additional 1.1 kg is gained every six weeks thereafter until term. The normal newborn weighs approximately 3,200 g at term. The newborn’s weight is dependent on its rate of growth and its gestational age at delivery. An infant that is growth retarded is said to be small for gestational age (SGA). If an infant is delivered prior to term it will have a reduced weight but will not necessarily be growth retarded. Factors associated with a preterm delivery are discussed elsewhere, and the focus of this discussion is on the growth-retarded newborn. The terms SGA and LBW will be used interchangeably. A low birth-weight infant is defined as an infant weighing less than 2,500 g, a very low birth weight is defined as less than 1,500 g, and extremely low birth weight is one that is less than 1,000 g (WHO 1969).
When examining causes of reduced growth, it is important to distinguish between asymmetrical and symmetrical growth retardation. Asymmetrical growth retardation, i.e., where the weight is affected more than the skeletal structure, is primarily associated with a risk factor operating during late pregnancy. On the other hand, symmetrical growth retardation may more likely be associated with an aetiology that operates over the entire period of gestation (Kline, Stein and Susser 1989). The difference in rates between asymmetrical and symmetrical growth retardation is especially apparent when comparing developing and developed countries. The rate of growth retardation in developing countries is 10 to 43%, and is primarily symmetrical, with the most important risk factor being poor nourishment. In developed countries foetal growth retardation is usually much lower, 3 to 8%, and is generally asymmetrical with a multifactorial aetiology. Hence, worldwide, the proportion of low birth-weight infants defined as intrauterine growth retarded rather than preterm varies dramatically. In Sweden and the United States, the proportion is approximately 45%, while in developing countries, such as India, the proportion varies between approximately 79 and 96% (Villar and Belizan 1982).
Studies of the Dutch famine showed that starvation confined to the third trimester depressed foetal growth in an asymmetric pattern, with birth weight being primarily affected and head circumference least affected (Stein, Susser and Saenger 1975). Asymmetry of growth also has been observed in studies of environmental exposures. In a study of 202 expectant mothers residing in neighbourhoods at high risk for lead exposures, prenatal maternal blood samples were collected between the sixth and the 28th week of gestation (Bornschein, Grote and Mitchell 1989). Blood lead levels were associated with both a decreased birth weight and length, but not head circumference, after adjustment for other relevant risk factors including length of gestation, socioeconomic status and use of alcohol or cigarettes. The finding of maternal blood lead as a factor in birth length was seen entirely in Caucasian infants. The birth length of Caucasian infants decreased approximately 2.5 cm per log unit increment in maternal blood lead. Care should be given to selection of the outcome variable. If only birth weight had been selected for study, the finding of the effects of lead on other growth parameters might have been missed. Also, if Caucasians and African Americans had been pooled in the above analysis, the differential effects on Caucasians, perhaps due to genetic differences in the storage and binding capacity of lead, may have been missed. A significant confounding effect also was observed between prenatal blood lead and maternal age and the birth weight of the offspring after adjustment for other covariables. The findings indicate that for a 30-year-old woman with an estimated blood lead level of about 20 mg/dl, the offspring weighed proximately 2,500 g compared with proximately 3,000 g for a 20-year-old with similar lead levels. The investigators speculated that this observed difference may indicate that older women are more sensitive to the additional insult of lead exposure or that older women may have had higher total lead burden from greater numbers of years of exposure or higher ambient lead levels when they were children. Another factor may be increased blood pressure. Nonetheless, the important lesson is that careful examination of high-risk subpopulations by age, race, economic status, daily living habits, sex of the offspring and other genetic differences may be necessary in order to discover the more subtle effects of exposures on foetal growth and development.
Risk factors associated with low birth weight are summarized in Table 5. Social class as measured by income or education persists as a risk factor in situations in which there are no ethnic differences. Other factors that may be operating under social class or race may include cigarette smoking, physical work, prenatal care and nutrition. Women between the ages of 25 and 29 are least likely to deliver a growth-retarded offspring. Maternal smoking increases the risk of low birth-weight offspring by about 200% for heavy smokers. Maternal medical conditions associated with LBW include placental abnormalities, heart disease, viral pneumonia, liver disease, re-eclamsia, eclamsia, chronic hypertension, weight gain and hyeremesis. An adverse pregnancy history of foetal loss, preterm delivery or prior LBW infant increases the risk of a current preterm low birth-weight infant two- to fourfold. An interval between births of less than a year triples the risk of having a low birth-weight offspring. Chromosomal anomalies associated with abnormal growth include Down’s syndrome, trisomy 18 and most malformation syndromes.
Smoking cigarettes is one of the primary behaviours most directly linked with lower weight offspring. Maternal smoking during pregnancy has been shown to increase the risk of a low birth-weight offspring two to three times and to cause an overall weight deficit of between 150 and 400 g. Nicotine and carbon monoxide are considered the most likely causative agents since both are rapidly and referentially transferred across the placenta. Nicotine is a powerful vasoconstrictor, and significant differences in the size of umbilical vessels of smoking mothers have been demonstrated. Carbon monoxide levels in cigarette smoke range from 20,000 to 60,000 m. Carbon monoxide has an affinity for haemoglobin 210 times that of oxygen, and because of lower arterial oxygen tension the foetus is especially compromised. Others have suggested that these effects are not due to smoking but are attributable to characteristics of smokers. Certainly occupations with potential carbon monoxide exposure, such as those associated with ul and aer, blast furnaces, acetylene, breweries, carbon black, coke ovens, garages, organic chemical synthesizers and petroleum refineries should be considered possible high risk occupations for pregnant employees.
Ethanol is also a widely used and researched agent associated with foetal growth retardation (as well as congenital anomalies). In a prospective study of 9,236 births, it was found that maternal alcohol consumption of more than 1.6 oz per day was associated with an increase in stillbirths and growth-retarded infants (Kaminski, Rumeau and Schwartz 1978). Smaller infant length and head circumference also are related to maternal alcohol ingestion.
In evaluating the possible effects of exposures on birth weight, some problematic issues must be considered. preterm delivery should be considered as a possible mediating outcome and the potential effects on gestational age considered. In addition, pregnancies having longer gestational length also have a longer opportunity for exposure. If enough women work late in pregnancy, the longest cumulative exposure may be associated with the oldest gestational ages and heaviest babies purely as an artifact. There are a number of procedures that can be used to overcome this problem including a variant of the Cox life-table regression model, which has the ability to handle time-dependent covariables.
Another problem centres on how to define lowered birth weight. Often studies define lower birth weight as a dichotomous variable, less than 2,500 g. The exposure, however, must have a very powerful effect in order to produce a drastic drop in the infant’s weight. Birth weight defined as a continuous variable and analysed in a multiple regression model is more sensitive for detecting subtle effects. The relative paucity of significant findings in the literature in relationship to occupational exposures and SGA infants may, in art, be caused by ignoring these design and analysis issues.
Conclusions
Studies of adverse pregnancy outcomes must characterize exposures during a fairly narrow window of time. If the woman has been transferred to another job or laid off work during a critical period of time such as organogenesis, the exposure-effect relationship can be severely altered. Therefore, the investigator is held to a high standard of identifying the woman’s exposure during a critical small time period as compared with other studies of chronic diseases, where errors of a few months or even years may have minimal impact.
Uterine growth retardation, congenital anomaly and spontaneous abortions are frequently evaluated in occupational exposure studies. There is more than one approach available to assess each outcome. These end-points are of public health importance due to both the psychological and the financial costs. Generally, nonsecificity in the exposure-outcome relationships has been observed, e.g., with exposure to lead, anaesthetic gases and solvents. Because of the potential for nonsecificity in the exposure-effect relationship, studies should be designed to assess several end-points associated with a range of possible mechanisms.