The major effects of high altitude on humans relate to the changes in barometric pressure (PB) and its consequential changes in the ambient pressure of oxygen (O2). Barometric pressure decreases with increasing altitude in a logarithmic fashion and can be estimated by the following equation:
![]()
where a = altitude, expressed in metres. In addition, the relationship of barometric pressure to altitude is influenced by other factors such as distance from the equator and season. West and Lahiri (1984) found that direct measurements of barometric pressure near the equator and at the summit of Mt. Everest (8,848 m) were greater than predictions based on the International Civil Aviation Organization Standard Atmosphere. Weather and temperature also affect the relationship between barometric pressure and altitude to the extent that a low-pressure weather system can reduce pressure, making sojourners to high altitude “physiologically higher”. Since the inspired partial pressure of oxygen (PO2) remains constant at approximately 20.93% of barometric pressure, the most important determinant of inspired PO2 at any altitude is the barometric pressure. Thus, inspired oxygen decreases with increasing altitude due to decreased barometric pressure, as shown in figure 1.
Figure 1. Effects of altitude on barometric pressure and inspired PO2

Temperature and ultraviolet radiation also change at high altitudes. Temperature decreases with increasing altitude at a rate of approximately 6.5 °C per 1,000 m. Ultraviolet radiation increases approximately 4% per 300 m due to decreased cloudiness, dust, and water vapour. In addition, as much as 75% of ultraviolet radiation can be reflected back by snow, further increasing exposure at high altitude. Survival in high altitude environments is dependent on adaptation to and/or protection from each of these elements.
Acclimatization
While rapid ascent to high altitudes often results in death, slow ascent by mountaineers can be successful when accompanied by compensatory physiological adaptation measures. Acclimatization to high altitudes is geared towards maintaining an adequate supply of oxygen to meet metabolic demands despite the decreasing inspired PO2. In order to achieve this goal, changes occur in all organ systems involved with oxygen uptake into the body, distribution of O2 to the necessary organs, and O2 unloading to the tissues.
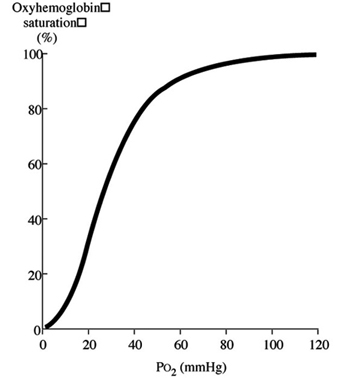
Discussion of oxygen uptake and distribution requires understanding the determinants of oxygen content in the blood. As air enters the alveolus, the inspired PO2 decreases to a new level (called the alveolar PO2) because of two factors: increased partial pressure of water vapour from humidification of inspired air, and increased partial pressure of carbon dioxide (PCO2) from CO2 excretion. From the alveolus, oxygen diffuses across the alveolar capillary membrane into the blood as a result of a gradient between alveolar PO2 and blood PO2. The majority of oxygen found in blood is bound to haemoglobin (oxyhaemoglobin). Thus, oxygen content is directly related to both the haemoglobin concentration in the blood and the percentage of O2 binding sites on haemoglobin that are saturated with oxygen (oxyhaemoglobin saturation). Therefore, understanding the relationship between arterial PO2 and oxyhaemoglobin saturation is essential for understanding the determinants of oxygen content in the blood. Figure 2 illustrates the oxyhaemoglobin dissociation curve. With increasing altitude, inspired PO2 decreases and, therefore, arterial PO2 and oxyhaemoglobin saturation decreases. In normal subjects, altitudes greater than 3,000 m are associated with sufficiently decreased arterial PO2 that oxyhaemoglobin saturation falls below 90%, on the steep portion of the oxyhaemoglobin dissociation curve. Further increases in altitude will predictably result in significant desaturation in the absence of compensatory mechanisms.
Figure 2. Oxyhaemoglobin dissociation curve
The ventilatory adaptations that occur in high-altitude environments protect the arterial partial pressure of oxygen against the effects of decreasing ambient oxygen levels, and can be divided into acute, subacute and chronic changes. Acute ascent to high altitude results in a fall in the inspired PO2 which in turn leads to a decrease in the arterial PO2 (hypoxia). In order to minimize the effects of decreased inspired PO2 on arterial oxyhaemoglobin saturation, the hypoxia that occurs at high altitude triggers an increase in ventilation, mediated through the carotid body (hypoxic ventilatory response–HVR). Hyperventilation increases carbon dioxide excretion and subsequently the arterial and then the alveolar partial pressure of carbon dioxide (PCO2) falls. The fall in alveolar PCO2 allows alveolar PO2 to rise, and consequently, arterial PO2 and arterial O2 content increases. However, the increased carbon dioxide excretion also causes a decrease in blood hydrogen ion concentration ([H+]) leading to the development of alkalosis. The ensuing alkalosis inhibits the hypoxic ventilatory response. Thus, on acute ascent to high altitude there is an abrupt increase in ventilation that is modulated by the development of an alkalosis in the blood.
Over the next several days at high altitude, further changes in ventilation occur, commonly referred to as ventilatory acclimatization. Ventilation continues to increase over the next several weeks. This further increase in ventilation occurs as the kidney compensates for the acute alkalosis by excretion of bicarbonate ions, with a resultant rise in blood [H+]. It was initially believed that renal compensation for the alkalosis removed the inhibitory influence of alkalosis on the hypoxic ventilatory response, thereby allowing the full potential of the HVR to be reached. However, measurements of blood pH revealed that the alkalosis persists despite the increase in ventilation. Other postulated mechanisms include: (1) cerebrospinal fluid (CSF) pH surrounding the respiratory control centre in the medulla may have returned to normal despite the persistent serum alkalosis; (2) increased sensitivity of the carotid body to hypoxia; (3) increased response of the respiratory controller to CO2. Once ventilatory acclimatization has occurred, both hyperventilation and the increased HVR persist for several days after return to lower altitudes, despite resolution of hypoxia.
Further ventilatory changes occur after several years of living at high altitude. Measurements in high-altitude natives have shown a decreased HVR when compared to values obtained in acclimatized individuals, although not to levels seen in subjects at sea level. The mechanism for the decreased HVR is unknown, but may be related to hypertrophy of the carotid body and/or development of other adaptive mechanisms for preserving tissue oxygenation such as: increased capillary density; increased gas exchange capacity of the tissues; increased number and density of mitochondria; or increased vital capacity.
In addition to its effect on ventilation, hypoxia also induces constriction of the vascular smooth muscle in the pulmonary arteries (hypoxic vasoconstriction). The ensuing increase in pulmonary vascular resistance and pulmonary artery pressure redirects blood flow away from poorly ventilated alveoli with low alveolar PO2 and towards better ventilated alveoli. In this manner, pulmonary arterial perfusion is matched to lung units that are well ventilated, providing another mechanism for preserving arterial PO2.
Oxygen delivery to the tissues is further enhanced by adaptations in the cardiovascular and haematological systems. On initial ascent to high altitude, heart rate increases, resulting in an increase in cardiac output. Over several days, cardiac output falls due to decreased plasma volume, caused by an increased water loss that occurs at high altitudes. With more time, increased erythropoietin production leads to increased haemoglobin concentration, providing the blood with increased oxygen-carrying capacity. In addition to increasing levels of haemoglobin, changes in the avidity of oxygen binding to haemoglobin may also help maintain tissue oxygenation. A shift of the oxyhaemoglobin dissociation curve to the right may be expected because it would favour release of oxygen to the tissues. However, data obtained from the summit of Mt. Everest and from hypobaric chamber experiments simulating the summit suggest that the curve is shifted to the left (West and Lahiri 1984; West and Wagner 1980; West et al. 1983). Although a left shift would make oxygen unloading to the tissues more difficult, it may be advantageous at extreme altitudes because it would facilitate oxygen uptake in the lungs despite markedly reduced inspired PO2 (43 mmHg on the summit of Mt. Everest versus 149 mmHg at sea level).
The last link in the chain of oxygen supply to the tissues is cellular uptake and utilization of O2. Theoretically, there are two potential adaptations that can occur. First, minimization of the distance that oxygen has to travel on diffusion out of the blood vessel and into the intracellular site responsible for oxidative metabolism, the mitochondria. Second, biochemical alterations can occur that improve mitochondrial function. Minimization of diffusion distance has been suggested by studies that show either increased capillary density or increased mitochondrial density in muscle tissue. It is unclear whether these changes reflect either recruitment or development of capillaries and mitochondria, or are an artefact due to muscle atrophy. In either case, the distance between the capillaries and the mitochondria would be decreased, thereby facilitating oxygen diffusion. Biochemical alterations that may improve mitochondrial function include increased myoglobin levels. Myoglobin is an intracellular protein that binds oxygen at low tissue PO2 levels and facilitates oxygen diffusion into the mitochondria. Myoglobin concentration increases with training and correlates with muscle cell aerobic capacity. Although these adaptations are theoretically beneficial, conclusive evidence is lacking.
Early accounts of high altitude explorers describe changes in cerebral function. Decreased motor, sensory and cognitive abilities, including decreased ability to learn new tasks and difficulty expressing information verbally, have all been described. These deficits may lead to poor judgement and to irritability, further compounding the problems encountered in high-altitude environments. On return to sea level, these deficits improve with a variable time course; reports have indicated impaired memory and concentration lasting from days to months, and decreased finger-tapping speed for one year (Hornbein et al. 1989). Individuals with greater HVR are more susceptible to long-lasting deficits, possibly because the benefit of hyperventilation on arterial oxyhaemoglobin saturation may be offset by hypocapnia (decreased PCO2 in the blood), which causes constriction of the cerebral blood vessels leading to decreased cerebral blood flow.
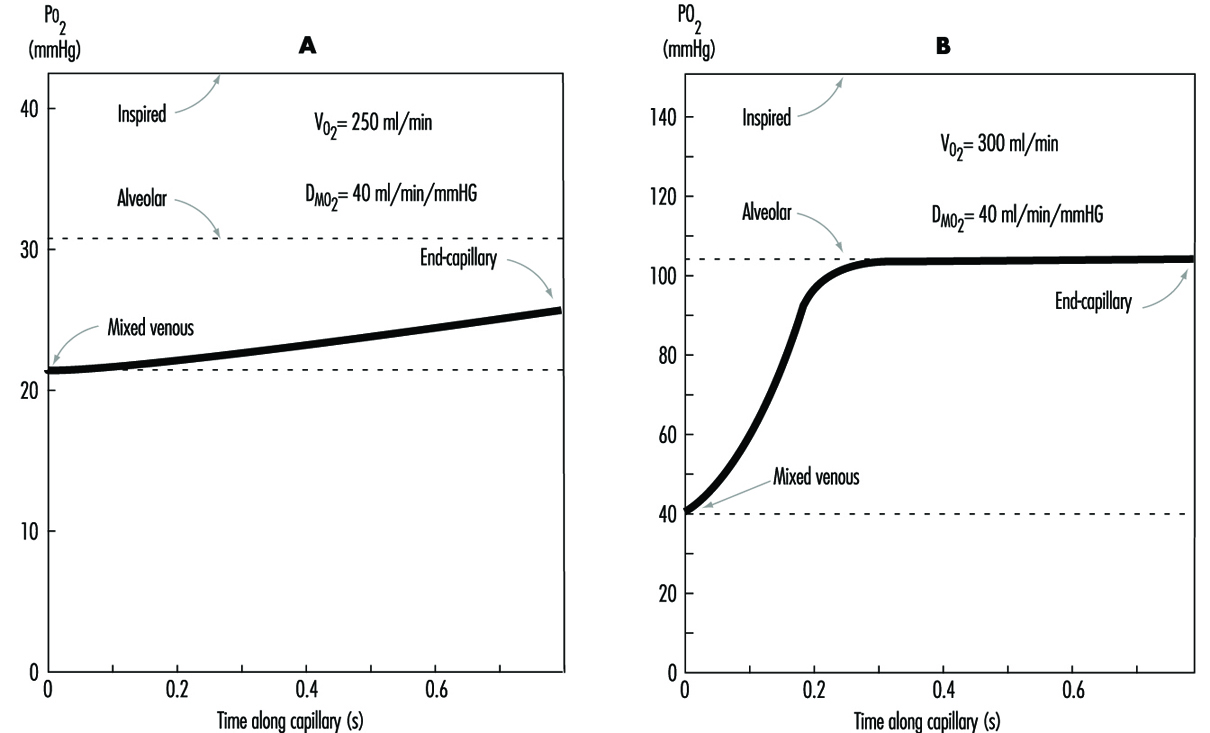
The preceding discussion has been limited to resting conditions; exercise provides an additional stress as oxygen demand and consumption increases. The fall in ambient oxygen at high altitude causes a fall in maximal oxygen uptake and, therefore, maximal exercise. In addition, the decreased inspired PO2 at high altitudes severely impairs oxygen diffusion into the blood. This is illustrated in figure 3, which plots the time course of oxygen diffusion into the alveolar capillaries. At sea level, there is excess time for equilibration of end-capillary PO2 to alveolar PO2, whereas at the summit of Mt. Everest, full equilibration is not realized. This difference is due to the decreased ambient oxygen level at high altitudes leading to a decreased diffusion gradient between alveolar and venous PO2. With exercise, cardiac output and blood flow increase, thereby reducing transit time of blood cells across the alveolar capillary, further exacerbating the problem. From this discussion, it becomes apparent that the left shift in the O2 and haemoglobin dissociation curve with altitude is necessary as compensation for the decreased diffusion gradient for oxygen in the alveolus.
Figure 3. The calculated time course of oxygen tension in the alveolar capillary
Disturbed sleep is common among sojourners at high altitude. Periodic (Cheyne-Stokes) breathing is universal and characterized by periods of rapid respiratory rate (hyperpnoea) alternating with periods of absent respirations (apnoea) leading to hypoxia. Periodic breathing tends to be more pronounced in individuals with the greatest hypoxic ventilatory sensitivity. Accordingly, sojourners with lower HVR have less severe periodic breathing. However, sustained periods of hypoventilation are then seen, corresponding with sustained decreases in oxyhaemoglobin saturation. The mechanism for periodic breathing probably relates to increased HVR causing increased ventilation in response to hypoxia. The increased ventilation leads to increased blood pH (alkalosis), which in turn suppresses ventilation. As acclimatization progresses, periodic breathing improves. Treatment with acetazolamide reduces periodic breathing and improves arterial oxyhaemoglobin saturation during sleep. Caution should be used with medications and alcohol that suppress ventilation, as they may exacerbate the hypoxia seen during sleep.
Pathophysiological Effects of Reduced Barometric Pressure
The complexity of human physiological adaptation to high altitude provides numerous potential maladaptive responses. Although each syndrome will be described separately, there is considerable overlap between them. Illnesses such as acute hypoxia, acute mountain sickness, high-altitude pulmonary oedema, and high-altitude cerebral oedema most likely represent a spectrum of abnormalities that share a similar pathophysiology.
Hypoxia
Hypoxia occurs with ascent to high altitudes because of the decreased barometric pressure and the resultant decrease in ambient oxygen. With rapid ascent, hypoxia occurs acutely, and the body does not have time to adjust. Mountaineers have generally been protected from the effects of acute hypoxia because of the time that elapses, and hence the acclimatization that occurs, during the climb. Acute hypoxia is problematic for both aviators and rescue personnel in high-altitude environments. Acute oxyhaemoglobin desaturation to values less than 40 to 60% leads to loss of consciousness. With less severe desaturation, individuals note headache, confusion, drowsiness and loss of coordination. Hypoxia also induces a state of euphoria which Tissandier, during his balloon flight in 1875, described as experiencing “inner joy”. With more severe desaturation, death occurs. Acute hypoxia responds rapidly and completely to either administration of oxygen or descent.
Acute mountain sickness
Acute mountain sickness (AMS) is the most common disorder in high-altitude environments and afflicts up to two-thirds of sojourners. The incidence of acute mountain sickness is dependent on multiple factors, including rate of ascent, length of exposure, degree of activity, and individual susceptibility. Identification of affected individuals is important in order to prevent progression to pulmonary or cerebral oedema. Identification of acute mountain sickness is made through recognition of characteristic signs and symptoms occurring in the appropriate setting. Most often, acute mountain sickness occurs within a few hours of a rapid ascent to altitudes greater than 2,500 m. The most common symptoms include headache that is more pronounced at night, loss of appetite that may be accompanied by nausea and vomiting, disturbed sleep, and fatigue. Individuals with AMS often complain of shortness of breath, cough and neurological symptoms such as memory deficits and auditory or visual disturbances. Findings on physical exam may be lacking, although fluid retention may be an early sign. The pathogenesis of acute mountain illness may relate to relative hypoventilation that would increase cerebral blood flow and intracranial pressure by increasing arterial PCO2 and decreasing arterial PO2. This mechanism may explain why persons with greater HVR are less likely to develop acute mountain sickness. The mechanism for fluid retention is not well understood, but may be related to abnormal plasma levels for proteins and/or hormones that regulate renal excretion of water; these regulators may respond to the increased activity of the sympathetic nervous system noted in patients with acute mountain sickness. The accumulation of water may in turn lead to the development of oedema or swelling of the interstitial spaces in the lungs. More severe cases may go on to develop pulmonary or cerebral oedema.
Prevention of acute mountain sickness can be accomplished through slow, graded ascent, allowing adequate time for acclimatization. This may be especially important for those individuals with greater susceptibility or a prior history of acute mountain sickness. In addition, administration of acetazolamide before or during ascent may help prevent and ameliorate symptoms of acute mountain sickness. Acetazolamide inhibits the action of carbonic anhydrase in the kidneys and leads to increased excretion of bicarbonate ions and water, producing an acidosis in the blood. The acidosis stimulates respiration, leading to increased arterial oxyhaemoglobin saturation and decreased periodic breathing during sleep. Through this mechanism, acetazolamide speeds the natural process of acclimatization.
Treatment of acute mountain sickness can be accomplished most effectively by descent. Further ascent to high altitudes is contra-indicated, as the disease may progress. When descent is not possible, oxygen may be administered. Alternatively, portable lightweight fabric hyperbaric chambers may be brought on expeditions to high-altitude environments. Hyperbaric bags are particularly valuable when oxygen is not available and descent is not possible. Several drugs are available that improve symptoms of acute mountain sickness, including acetazolamide and dexamethasone. The mechanism of action of dexamethasone is unclear, although it may act by decreasing oedema formation.
High-altitude pulmonary oedema
High-altitude pulmonary oedema affects approximately 0.5 to 2.0% of individuals who ascend to altitudes greater than 2,700 m and is the most common cause of death due to illnesses encountered at high altitudes. High-altitude pulmonary oedema develops from 6 to 96 hours after ascent. Risk factors for the development of high-altitude pulmonary oedema are similar to those for acute mountain sickness. Common early signs include symptoms of acute mountain sickness accompanied by decreased exercise tolerance, increased recovery time after exercise, shortness of breath on exertion, and persistent dry cough. As the condition worsens, the patient develops shortness of breath at rest, findings of audible congestion in the lungs, and cyanosis of the nail beds and lips. The pathogenesis of this disorder is uncertain but probably relates to increased microvascular pressure or increased permeability of the microvasculature leading to the development of pulmonary oedema. Although pulmonary hypertension may help explain the pathogenesis, elevation in the pulmonary artery pressure due to hypoxia has been observed in all individuals who ascend to high altitude, including those who do not develop pulmonary oedema. Nevertheless, susceptible individuals may possess uneven hypoxic constriction of the pul-monary arteries, leading to over-perfusion of the microvasculature in localized areas where hypoxic vasoconstriction was absent or diminished. The resulting increase in pressure and shear forces may damage the capillary membrane, leading to oedema formation. This mechanism explains the patchy nature of this disease and its appearance on x-ray examination of the lungs. As with acute mountain sickness, individuals with a lower HVR are more likely to develop high-altitude pulmonary oedema as they have lower oxyhaemoglobin saturations and, therefore, greater hypoxic pulmonary vasoconstriction.
Prevention of high-altitude pulmonary oedema is similar to prevention of acute mountain sickness and includes gradual ascent and use of acetazolamide. Recently, use of the smooth-muscle relaxing agent nifedipine has been shown to be of benefit in preventing disease in individuals with a prior history of high-altitude pulmonary oedema. Additionally, avoidance of exercise may have a preventive role, although it is probably limited to those individuals who already posses a subclinical degree of this disease.
Treatment of high-altitude pulmonary oedema is best accomplished by assisted evacuation to a lower altitude, keeping in mind that the victim needs to limit his or her exertion. After descent, improvement is rapid and additional treatment other than bed rest and oxygen are usually not necessary. When descent is not possible, oxygen therapy may be beneficial. Drug treatment has been attempted with multiple agents, most successfully with the diuretic furosemide and with morphine. Caution must be used with these drugs, as they can lead to dehydration, decreased blood pressure, and respiratory depression. Despite the effectiveness of descent as therapy, mortality remains at approximately 11%. This high mortality rate may reflect failure to diagnose the disease early in its course, or inability to descend coupled with lack of availability of other treatments.
High-altitude cerebral oedema
High-altitude cerebral oedema represents an extreme form of acute mountain sickness that has progressed to include generalized cerebral dysfunction. The incidence of cerebral oedema is unclear because it is difficult to differentiate a severe case of acute mountain sickness from a mild case of cerebral oedema. The pathogenesis of high-altitude cerebral oedema is an extension of the pathogenesis of acute mountain sickness; hypoventilation increases cerebral blood flow and intracranial pressure progressing to cerebral oedema. Early symptoms of cerebral oedema are identical to symptoms of acute mountain sickness. As the disease progresses, additional neurological symptoms are noted, including severe irritability and insomnia, ataxia, hallucinations, paralysis, seizures and eventually coma. Examination of the eyes commonly reveals swelling of the optic disc or papilloedema. Retinal haemorrhages are frequently noted. In addition, many cases of cerebral oedema have concurrent pulmonary oedema.
Treatment of high-altitude cerebral oedema is similar to treatment of other high-altitude disorders, with descent being the preferred therapy. Oxygen should be administered to maintain oxyhaemoglobin saturation greater that 90%. Oedema formation may be decreased with use of corticosteroids such as dexamethasone. Diuretic agents have also been utilized to decrease oedema, with uncertain efficacy. Comatose patients may require additional support with airway management. The response to treatment is variable, with neurological deficits and coma persisting for days to weeks after evacuation to lower altitudes. Preventative measures for cerebral oedema are identical to measures for other high-altitude syndromes.
Retinal haemorrhages
Retinal haemorrhages are extremely common, affecting up to 40% of individuals at 3,700 m and 56% at 5,350 m. Retinal haemorrhages are usually asymptomatic. They are most likely caused by increased retinal blood flow and vascular dilatation due to arterial hypoxia. Retinal haemorrhages are more common in individuals with headaches and can be precipitated by strenuous exercise. Unlike other high-altitude syndromes, retinal haemorrhages are not preventable by acetazolamide or furosemide therapy. Spontaneous resolution is usually seen within two weeks.
Chronic mountain sickness
Chronic mountain sickness (CMS) afflicts residents and long-term inhabitants of high altitude. The first description of chronic mountain sickness reflected Monge’s observations of Andean natives living at altitudes above 4,000 m. Chronic mountain sickness, or Monge’s disease, has since been described in most high-altitude dwellers except Sherpas. Males are more commonly affected than females. Chronic mountain sickness is characterized by plethora, cyanosis and elevated red blood cell mass leading to neurological symptoms that include headache, dizziness, lethargy and impaired memory. Victims of chronic mountain sickness may develop right heart failure, also called cor pulmonale, due to pulmonary hypertension and markedly reduced oxyhaemoglobin saturation. The pathogenesis of chronic mountain sickness is unclear. Measurements from affected individuals have revealed a decreased hypoxic ventilatory response, severe hypoxemia that is exacerbated during sleep, increased haemoglobin concentration and increased pulmonary artery pressure. Although a cause-and-effect relationship seems likely, evidence is lacking and often confusing.
Many symptoms of chronic mountain sickness can be ameliorated by descent to sea level. Relocation to sea level removes the hypoxic stimulus for red blood cell production and pulmonary vasoconstriction. Alternate treatments include: phlebotomy to reduce red blood cell mass, and low-flow oxygen during sleep to improve hypoxia. Therapy with medroxyprogesterone, a respiratory stimulant, has also been found to be effective. In one study, ten weeks of medroxyprogesterone therapy was followed by improved ventilation and hypoxia, and decreased red blood cell counts.
Other conditions
Patients with sickle cell disease are more likely to suffer from painful vaso-occlusive crisis at high altitude. Even moderate altitudes of 1,500 m have been known to precipitate crises, and altitudes of 1,925 m are associated with a 60% risk of crises. Patients with sickle cell disease residing at 3,050 m in Saudi Arabia have twice as many crises as patients residing at sea level. In addition, patients with sickle cell trait may develop splenic infarct syndrome on ascent to high altitude. Likely aetiologies for the increased risk of vaso-occlusive crisis include: dehydration, increased red blood cell count, and immobility. Treatment of vaso-occlusive crisis includes descent to sea level, oxygen and intravenous hydration.
Essentially no data exist describing the risk to pregnant patients on ascent to high altitudes. Although patients residing at high altitude have an increased risk of pregnancy-induced hypertension, no reports of increased foetal demise exist. Severe hypoxia may cause abnormalities in foetal heart rate; however, this occurs only at extreme altitudes or in the presence of high-altitude pulmonary oedema. Therefore, the greatest risk to the pregnant patient may relate to the remoteness of the area rather than to altitude-induced complications.