- You are here:
-
Home

-
Part XVII. Services and Trade
-
Health Care Facilities and Services
- The Physical Environment and Health Care
Coal Workers' Lung Diseases
Coal miners are subject to a number of lung diseases and disorders arising from their exposure to coal mine dust. These include pneumoconiosis, chronic bronchitis and obstructive lung disease. The occurrence and severity of disease depends on the intensity and duration of dust exposure. The specific composition of the coal mine dust also has a bearing on some health outcomes.
In the developed countries, where high prevalences of lung disease existed in the past, reductions in dust levels brought about by regulation have led to substantial drops in disease prevalence since the 1970s. In addition, major reductions in the mining work force in most of those countries over recent decades, partly brought about by changes in technology and resulting improvements in productivity, will result in further reductions in overall disease levels. Miners in other countries, where coal mining is a more recent phenomenon and dust controls are less aggressive, have not been so fortunate. This problem is exacerbated by the high cost of modern mining technology, forcing the employment of large numbers of workers, many of whom are at high risk of disease development.
In the following text, each disease or disorder is considered in turn. Those specific to coal mining, such as coal workers’ pneumoconiosis are described in detail; the description of others, such as obstructive lung disease, is restricted to those aspects that relate to coal miners and dust exposure.
Coal Workers’ Pneumoconiosis
Coal workers’ pneumoconiosis (CWP) is the disease most commonly associated with coal mining. It is not a fast-developing disease, usually taking at least ten years to be manifested, and often much longer when exposures are low. In its initial stages it is an indicator of excessive lung dust retention, and may be associated with few symptoms or signs in itself. However, as it advances, it puts the miner at increasing risk of development of the much more serious progressive massive fibrosis (PMF).
Pathology
The classic lesion of CWP is the coal macule, a collection of dust and dust-laden macrophages around the periphery of the respiratory bronchioles. The macules contain minimal collagen and are thus usually not palpable. They are about 1 to 5 mm in size, and are frequently accompanied by an enlargement of the adjacent air spaces, termed focal emphysema. Though often very numerous, they are not usually evident on a chest radiograph.
Another lesion associated with CWP is the coal nodule. These larger lesions are palpable and contain a mixture of dust-laden macrophages, collagen and reticulin. The presence of coal nodules, with or without silicotic nodules (see below), indicates lung fibrosis, and is largely responsible for the opacities seen on chest radiographs. Macronodules (7 to 20 mm) in size may coalesce to form progressive massive fibrosis (see below), or PMF may develop from a single macronodule.
Silicotic nodules (described under silicosis) have been found in a significant minority of underground coal miners. For most, the cause may rest simply with the silica present in the coal dust, although exposure to pure silica in some jobs is certainly an important factor (e.g., among surface drillers, underground motormen and roof bolters).
Radiography
The most useful indicator of CWP in miners during life is obtained using the routine chest radiograph. Dust deposits and the nodular tissue reactions attenuate the x-ray beam and result in opacities on the film. The profusion of these opacities can be assessed systematically by using a standardized method of radiograph description such as that disseminated by the ILO and described else in this chapter. In this method, individual posterior-anterior films are compared to standard radiographs showing increasing profusion of small opacities, and the film classified into one of four major categories (0, 1, 2, 3) based on its similarity to the standard. A secondary classification is also made, depending on the reader’s assessment of the film’s similarity to adjacent ILO categories. Other aspects of the opacities, such as size, shape and region of occurrence in the lung are also noted. Some countries, such as China and Japan, have developed similar systems for systematic radiograph description or interpretation that are particularly suited to their own needs.
Traditionally, small rounded types of opacity have been associated with coal mining. However, more recent data indicate that irregular types can also result from exposure to coal mine dust. The opacities of CWP and silicosis are often indistinguishable on the radiograph. However, there is some evidence that larger sized opacities (type r) more often indicate silicosis.
It is important to note that a substantial amount of pathologic abnormality related to pneumoconiosis may be present in the lung before it can be detected on the routine chest x ray. This is particularly true for macular deposition, but it becomes progressively less true with greater profusion and size of nodules. Concomitant emphysema may also reduce the visibility of lesions on the chest x ray. Computerized tomography (CT)—particularly high-resolution computerized tomography (HRCT)—may permit visualization of abnormalities not clearly evident on routine chest x rays, although CT is not necessary for routine clinical diagnosis of miners’ lung diseases and is not indicated for medical surveillance of miners.
Clinical aspects
The development of CWP, although a marker of excessive lung dust retention, in itself is often unaccompanied by any overt clinical signs. This should not, however, be taken to imply that the inhalation of coal mine dust is without risk, for it is now well known that other lung diseases can arise from dust exposure. Pulmonary hypertension is more often noted in miners who develop airflow obstruction in association with CWP. Moreover, once CWP has developed, it usually progresses unless dust exposure ceases, and may progress thereafter. It also puts the miner at greatly increased risk of development of the clinically ominous PMF, with the likelihood of subsequent impairment, disability and premature mortality.
Disease mechanisms
Development of the earliest change of CWP, the dust macule, represents the effects of dust deposition and accumulation. The subsequent stage, that is, the development of nodules, results from the lung’s inflammatory and fibrotic reaction to the dust. In this, the roles of silica and non-silica dust have long been debated. On the one hand, silica dust is known to be considerably more toxic than coal dust. Yet, on the other hand, epidemiological studies have shown no strong evidence implicating silica exposure in CWP prevalence or incidence. Indeed, it seems that almost an inverse relationship exists, in that disease levels tend to be elevated where silica levels are lower (e.g., in areas where anthracite is mined). Recently, some understanding of this paradox has been gained through studies of particle characteristics. These studies indicate that not only the quantity of silica present in the dust (as measured conventionally using infrared spectrometry or x-ray diffraction), but also the bioavailability of the surface of the silica particles may be related to toxicity. For example, clay coating (occlusion) may play an important modifying role. Another important factor under current investigation concerns surface charge in the form of free radicals and the effects of “freshly fractured” versus “aged” silica-containing dusts.
Surveillance and epidemiology
The prevalence of CWP among underground miners varies with the kind of job, tenure and age. A recent study of US coal miners revealed that from 1970 to 1972 about 25 to 40% of working coal miners had category 1 or greater small rounded opacities after 30 or more years in mining. This prevalence reflects exposure to levels of 6 mg/m3 or more of respirable dust among coal face workers prior to that time. The introduction of a dust limit of 3 mg/m3 in 1969, with a reduction to 2 mg/m3 in 1972 has led to a decline in disease prevalence to about half of the former levels. Declines related to dust control have been noted elsewhere, for example, in the United Kingdom and Australia. Unfortunately, these gains have been counterbalanced by temporal increases in prevalence elsewhere.
An exposure-response relationship for prevalence or incidence of CWP and dust exposure has been demonstrated in a number of studies. These have shown that the primary significant dust exposure variable is exposure to mixed mine dust. Intensive studies by British researchers failed to disclose any major influence of silica exposure, as long as the percentage of silica was less than about 5%. Coal rank (percentage carbon) is another important predictor of CWP development. Studies in the United States, the United Kingdom, Germany and elsewhere have given clear indications that the prevalence and incidence of CWP increases markedly with coal rank, these being substantially greater where anthracite (high rank) coal is mined. No other environmental variables have been found to exert any major effects on CWP development. Miner age appears to have some bearing on disease development, since older miners appear to be at increased risk. However, it is not entirely clear whether this implies that older miners are more susceptible, whether it is a residence time effect, or is simply an artefact (the age effect might reflect underestimation of exposure estimates for older miners, for example). Cigarette smoking does not appear to increase the risk of CWP development.
Research in which miners were followed-up with chest radiographs every five years shows that the risk of developing PMF over the five years is clearly related to the category of CWP as revealed on the initial chest x ray. Since the risk at category 2 is much greater than that at category 1, conventional wisdom at one time was that miners should be prevented from reaching category 2 if at all possible. However, in most mines there are usually many more miners with category 1 CWP compared to category 2. Thus, the lower risk for category 1 compared to category 2 is offset somewhat by the larger numbers of miners with category 1. On this showing, it has become clear that all pneumoconiosis should be prevented.
Mortality
Miners as a group have been observed to have increased risk of death from non-malignant respiratory diseases, and there is evidence that the mortality among miners with CWP is somewhat increased over those of similar age without the disease. However, the effect is smaller than the excess seen for miners with PMF (see below).
Prevention
The only protection against CWP is minimization of dust exposure. If possible, this should be achieved by dust suppression methods, such as ventilation and water sprays, rather than by respirator use or administrative controls, for example, worker rotation. In this respect, there is now good evidence that regulatory actions in some countries to reduce the level of dust, taken around the 1970s, has resulted in greatly reduced levels of disease. Transfer of workers with early signs of CWP to less dusty jobs is a prudent action, although there is little practical evidence that such programmes have succeeded in preventing disease progression. For this reason, dust suppression must remain the primary method of disease prevention.
Ongoing, aggressive monitoring of dust exposure and the conscious exertion of control efforts can be supplemented by health screening surveillance of miners. If miners are found to develop dust-related diseases, efforts at exposure control should be intensified throughout the workplace and miners with dust effects should be offered work in low-dust areas of the mine environment.
Treatment
Although several forms of treatment have been tried, including aluminium powder inhalation, and the administration of tetrandine, no treatment is known that effectively reverses or slows the fibrotic process in the lung. Currently, primarily in China, but elsewhere also, whole-lung lavage is being tried with the intent of reducing the total lung dust burden. Although the procedure can result in the removal of a considerable amount of dust, its risks, benefits and role in the management of miners’ health are unclear.
In other respects, treatment should be directed towards preventing complications, maximizing the miners’ functional status and alleviating their symptoms, whether due to CWP or to other, concomitant respiratory diseases. In general, miners who develop dust-induced lung diseases should evaluate their current dust exposures and utilize the resources of government and labour organizations to find the avenues available to reduce all adverse respiratory exposures. For miners who smoke, smoking cessation is an initial step in personal exposure management. Prevention of infectious complications of chronic lung disease with available pneumococcal and yearly influenza vaccines is suggested. Early investigation of symptoms of lung infection, with particular attention to mycobacterial disease, is also recommended. The treatments for acute bronchitis, bronchospasm and congestive heart failure among miners are similar to those for patients without dust-related disease.
Progressive Massive Fibrosis
PMF, sometimes referred to as complicated pneumoconiosis, is diagnosed when one or more large fibrotic lesions (whose definition depends on the mode of detection) are present in one or both lungs. As its name implies, PMF often becomes more severe over time, even in the absence of additional dust exposure. It can also develop after dust exposure has ceased, and may often cause disability and premature mortality.
Pathology
PMF lesions may be unilateral or bilateral, and are most often found in the upper or middle lobes of the lung. The lesions are formed of collagen, reticulin, coal mine dust and dust-laden macrophages, while the centre may contain a black liquid which cavitates on occasion. US pathology standards require the lesions to be 2 cm in size or larger to be identified as PMF entities in surgical or autopsy specimens.
Radiology
Large opacities >>1 cm) on the radiograph, coupled with a history of extensive coal mine dust exposure, are taken to imply the presence of PMF. However, it is important that other diseases such as lung cancer, tuberculosis and granulomas be considered. Large opacities are usually seen on a background of small opacities, but development of PMF from a category 0 profusion has been noted over a five-year period.
Clinical aspects
Diagnostic possibilities for each individual miner with large chest opacities must be appropriately evaluated. Clinically stable miners with bilateral lesions in the typical upper-lung distribution and with pre-existing simple CWP may present little diagnostic challenge. However, miners with progressive symptoms, risk factors for other disorders (e.g., tuberculosis), or atypical clinical features should undergo a thorough appropriate examination before the diagnostician attributes the lesions to PMF.
Dyspnoea and other respiratory symptoms often accompany PMF, but may not necessarily be due to the disease itself. Congestive heart failure (due to pulmonary hypertension and cor pulmonale) is a not infrequent complication.
Disease mechanisms
Despite extensive research, the actual cause of PMF development remains unclear. Over the years, various hypotheses have been proposed, but none is fully satisfactory. One prominent theory was that tuberculosis played a role. Indeed, tuberculosis is often present in miners with PMF, particularly in the developing countries. However, PMF has been found to develop in miners in whom there was no sign of tuberculosis, and tuberculin reactivity has not been found to be elevated in miners with pneumoconiosis. Despite investigation, consistent evidence of the role of the immune system in PMF development is lacking.
Surveillance and epidemiology
As with CWP, PMF levels have been declining in countries which have strict dust control regulations and programmes. A recent study of US miners revealed that about 2% of coal miners working underground had PMF after 30 or more years in mining (although this figure may have been biased by affected miners leaving the work force).
Exposure-response investigations of PMF have shown that exposure to coal mine dust, category of CWP, coal rank and age are the primary determinants of disease development. As with CWP, epidemiological studies have found no major effect of silica dust. Although it was thought at one time that PMF developed only on a background of the small opacities of CWP, recently this has been found not to be the case. Miners with an initial chest x ray showing category 0 CWP have been shown to develop PMF over five years, with the risk increasing with their cumulative dust exposure. Also, miners may develop PMF after cessation of dust exposure.
Mortality
PMF leads to premature mortality, the prognosis worsening with increasing stage of the disease. A recent study showed that miners with category C PMF had only one-fourth the rate of survival over 22 years compared to miners with no pneumoconiosis. This effect was manifested over all age groups.
Prevention
Avoidance of dust exposure is the only way to prevent PMF. Since the risk of its development increases sharply with increasing category of simple CWP, a strategy for secondary prevention of PMF is for miners to undergo periodic chest x rays and to terminate or reduce their exposure if simple CWP is detected. Although this approach appears valid and has been adopted in certain jurisdictions, its effectiveness has not been evaluated systematically.
Treatment
There is no known treatment for PMF. Medical care should be organized around ameliorating the condition and associated lung illnesses, while protecting against infectious complications. Although maintaining functional stability may be more difficult in patients with PMF, in other respects, management is similar to simple CWP.
Obstructive Lung Disease
There is now consistent and convincing evidence of a relationship between lung function loss and dust exposure. Various studies in different countries have looked at the influence of dust exposure on absolute values of, and temporal changes in, measurements of ventilatory function, such as forced expiratory volume in one second (FEV1), forced vital capacity (FVC) and flow rates. All have found evidence that dust exposure leads to a reduction in lung function, and the results have been strikingly similar for several recent British and US investigations. These indicate that over the course of a year, dust exposure at the coal face brings about, on average, a reduction in lung function equivalent to smoking half a pack of cigarettes each day. The studies also demonstrate that effects vary, and a given miner may develop effects equal to, or worse than, those expected from cigarette smoking, particularly if the individual has experienced higher dust exposures.
The effects of dust exposure have been found in both those who have never smoked and in current smokers. Moreover, there is no evidence that smoking exacerbates the dust exposure effect. Rather, studies have generally shown a slightly smaller effect in current smokers, a result that may be due to healthy worker selection. It is important to note that the relationship between dust exposure and ventilatory decline appears to exist independently of pneumoconiosis. That is, it is not a requirement that pneumoconiosis be present for there to be reduced lung function. To the contrary, it appears rather that the inhaled dust can act along multiple pathways, leading to pneumoconiosis in some miners, to obstruction in others and to multiple outcomes in yet others. In contrast to miners with CWP alone, miners with respiratory symptoms have significantly lower lung function, after standardization for age, smoking, dust exposure and other factors.
Recent work on ventilatory function changes has involved the exploration of longitudinal changes. The results indicate that there may be a non-linear trend of decline over time in new miners, a high initial rate of loss being followed by a more moderate decline with continued exposure. Furthermore, there is evidence that miners who react to the dust may choose, if possible, to remove themselves from the heavier exposures.
Chronic Bronchitis
Respiratory symptoms, such as chronic cough and phlegm production, are a frequent consequence of work in coal mining, most studies showing an excess prevalence compared to non-exposed control groups. Moreover, the prevalence and incidence of respiratory symptoms has been shown to increase with cumulative dust exposure, after taking into account age and smoking. The presence of symptoms appears to be associated with a reduction in lung function over and above that due to dust exposure and other putative causes. This suggests that dust exposure may be instrumental in initiating certain disease processes that then progress regardless of further exposure. A relationship between bronchial gland size and dust exposure has been demonstrated pathologically, and it has been found that mortality from bronchitis and emphysema increases with increasing cumulative dust exposure.
Emphysema
Pathological studies have repeatedly found an excess of emphysema in coal miners compared to control groups. Moreover, the degree of emphysema has been found to be related both to the amount of dust in the lungs and to pathological assessments of pneumoconiosis. Furthermore, it is important to recognize that there is evidence that the presence of emphysema is related to dust exposure and to the percentage of predicted FEV1. Hence, these results are consistent with the view that dust exposure can lead to disability through causing emphysema.
The form of emphysema most clearly associated with coal mining is focal emphysema. This consists of zones of enlarged air spaces, 1 to 2 mm in size, adjacent to dust macules surrounding the respiratory bronchioles. The current thinking is that the emphysema is formed from tissue destruction, rather than from distension or dilation. Apart from focal emphysema, there is evidence that centriacinar emphysema has an occupational origin, and that total emphysema, (i.e., the extent of all types) is correlated with tenure in mining, in those who have never smoked as well as in smokers. There is no evidence that smoking potentiates the dust exposure/emphysema relationship. However, there are indications of an inverse relationship between the silica content of lungs and the presence of emphysema.
The issue of emphysema has long been controversial, with some stating that selection bias and smoking make interpretation of pathological studies difficult. In addition, some consider that focal emphysema has only trivial effects on lung function. However, pathological studies undertaken since the 1980s have been responsive to earlier criticisms, and indicate that the effect of dust exposure may be more significant for miners’ health than previously thought. This point of view is supported by recent findings that mortality from bronchitis and emphysema is related to cumulative dust exposure.
Silicosis
Silicosis, though associated more with industries other than coal mining, can occur in coal miners. In underground mines, it is found most frequently in workers in certain jobs where exposure to pure silica typically occurs. Such workers include roof bolters, who drill into the ceiling rock, which can often be sandstone or other rock with high silica content; motormen, drivers of rail transport who are exposed to the dust generated by sand placed on the tracks to lend traction; and rock drillers, who are involved in mine development. Rock drillers at surface coal mines have been shown to be at particular risk in the United States, with some developing acute silicosis after only a few years of exposure. Based on pathological evidence, as noted below, some degree of silicosis may afflict many more coal miners than just those working the jobs noted above.
Silicotic nodules in coal miners are similar in nature to those observed elsewhere, and consist of a whorled pattern of collagen and reticulin. One large autopsy study has revealed that about 13% of coal miners had silicotic nodules in their lungs. Although one job, (that of motorman) was notable for having a much higher prevalence of silicotic nodules (25%), there was little variation in the prevalence among miners in other jobs, suggesting that the silica in the mixed mine dust was responsible.
Silicosis cannot be reliably differentiated from coal workers’ pneumoconiosis on a radiograph. However, there is some evidence that the larger type of small opacities (type r) are indicative of silicosis.
Rheumatoid Pneumoconiosis
Rheumatoid pneumoconiosis, one variant of which is called Caplan’s syndrome, is the term used for a condition affecting dust-exposed workers who develop multiple large radiographic shadows. Pathologically, these lesions resemble rheumatoid nodules rather than PMF lesions, and often arise over a short time interval. Active arthritis or the presence of circulating rheumatoid factor are generally found, but occasionally are absent.
Lung Cancer
Included in the occupational exposures suffered by coal miners are a number of substances that are potential carcinogens. Some of these are silica and benzo(a)pyrenes. Yet, there is no clear evidence of an excess of deaths from lung cancer in coal miners. One obvious explanation for this is that coal miners are forbidden to smoke underground because of the danger of fires and explosions. However, the fact that no exposure-response relationship between lung cancer and dust exposure has been detected suggests that coal mine dust is not a major cause of lung cancer in the industry.
Regulatory Limits on Dust Exposure
The World Health Organization (WHO) has recommended a “tentative health-based exposure limit” for respirable coal mine dust (with less than 6% respirable quartz) ranging from 0.5 to 4 mg/m3. WHO suggests a 2 in 1,000 risk of PMF over a working lifetime as a criterion, and recommends that mine-based environmental factors, including coal rank, percentage of quartz and particle size should be taken into account when setting limits.
Currently, among the major coal-producing countries, limits are based on regulating coal dust alone (e.g., 3.8 mg/m3 in the United Kingdom, 5 mg/m3 in Australia and Canada) or on regulating a mixture of coal and silica as in the United States (2 mg/m3 when the per cent quartz is 5 or less, or (10 mg/m3)/per cent SiO2), or in Germany (4 mg/m3 when the per cent quartz is 5 or less, or 0.15 mg/m3 otherwise), or on regulating pure quartz (e.g., Poland, with a 0.05 mg/m3 limit).
Silicosis
Silicosis is a fibrotic disease of the lungs caused by the inhalation, retention and pulmonary reaction to crystalline silica. Despite knowledge of the cause of this disorder—respiratory exposures to silica containing dusts—this serious and potentially fatal occupational lung disease remains prevalent throughout the world. Silica, or silicon dioxide, is the predominant component of the earth’s crust. Occupational exposure to silica particles of respirable size (aerodynamic diameter of 0.5 to 5μm) is associated with mining, quarrying, drilling, tunnelling and abrasive blasting with quartz containing materials (sandblasting). Silica exposure also poses a hazard to stonecutters, and pottery, foundry, ground silica and refractory workers. Because crystalline silica exposure is so widespread and silica sand is an inexpensive and versatile component of many manufacturing processes, millions of workers throughout the world are at risk of the disease. The true prevalence of the disease is unknown.
Definition
Silicosis is an occupational lung disease attributable to the inhalation of silicon dioxide, commonly known as silica, in crystalline forms, usually as quartz, but also as other important crystalline forms of silica, for example, cristobalite and tridymite. These forms are also called “free silica” to distinguish them from the silicates. The silica content in different rock formations, such as sandstone, granite and slate, varies from 20 to nearly 100%.
Workers in High-Risk Occupations and Industries
Although silicosis is an ancient disease, new cases are still reported in both the developed and developing world. In the early part of this century, silicosis was a major cause of morbidity and mortality. Contemporary workers are still exposed to silica dust in a variety of occupations—and when new technology lacks adequate dust control, exposures may be to more hazardous dust levels and particles than in non-mechanized work settings. Whenever the earth’s crust is disturbed and silica-containing rock or sand is used or processed, there are potential respiratory risks for workers. Reports continue of silicosis from industries and work settings not previously recognized to be at risk, reflecting the nearly ubiquitous presence of silica. Indeed, due to the latency and chronicity of this disorder, including the development and progression of silicosis after exposure has ceased, some workers with current exposures may not manifest disease until the next century. In many countries throughout the world, mining, quarrying, tunnelling, abrasive blasting and foundry work continue to present major risks for silica exposure, and epidemics of silicosis continue to occur, even in developed nations.
Forms of Silicosis—Exposure History and Clinicopathologic Descriptions
Chronic, accelerated and acute forms of silicosis are commonly described. These clinical and pathologic expressions of the disease reflect differing exposure intensities, latency periods and natural histories. The chronic or classic form usually follows one or more decades of exposure to respirable dust containing quartz, and this may progress to progressive massive fibrosis (PMF). The accelerated form follows shorter and heavier exposures and progresses more rapidly. The acute form may occur after short-term, intense exposures to high levels of respirable dust with high silica content for periods that may be measured in months rather than years.
Chronic (or classic) silicosis may be asymptomatic or result in insidiously progressive exertional dyspnoea or cough (often mistakenly attributed to the ageing process). It presents as a radiographic abnormality with small (<10 mm), rounded opacities predominantly in the upper lobes. A history of 15 years or more since onset of exposure is common. The pathologic hallmark of the chronic form is the silicotic nodule. The lesion is characterized by a cell-free central area of concentrically arranged, whorled hyalinized collagen fibers, surrounded by cellular connective tissue with reticulin fibers. Chronic silicosis may progress to PMF (sometimes referred to as complicated silicosis), even after exposure to silica-containing dust has ceased.
Progressive massive fibrosis is more likely to present with exertional dyspnoea. This form of disease is characterized by nodular opacities greater than 1 cm on chest radiograph and commonly will involve reduced carbon monoxide diffusing capacity, reduced arterial oxygen tension at rest or with exercise, and marked restriction on spirometry or lung volume measurement. Distortion of the bronchial tree may also lead to airway obstruction and productive cough. Recurrent bacterial infection not unlike that seen in bronchiectasis may occur. Weight loss and cavitation of the large opacities should prompt concern for tuberculosis or other mycobacterial infection. Pneumothorax may be a life-threatening complication, since the fibrotic lung may be difficult to re-expand. Hypoxaemic respiratory failure with cor pulmonale is a common terminal event.
Accelerated silicosis may appear after more intense exposures of shorter (5 to 10 years) duration. Symptoms, radiographic findings and physiological measurements are similar to those seen in the chronic form. Deterioration in lung function is more rapid, and many workers with accelerated disease may develop mycobacterial infection. Auto-immune disease, including scleroderma or systemic sclerosis, is seen with silicosis, often of the accelerated type. The progression of radiographic abnormalities and functional impairment can be very rapid when auto-immune disease is associated with silicosis.
Acute silicosis may develop within a few months to 2 years of massive silica exposure. Dramatic dyspnoea, weakness, and weight loss are often presenting symptoms. The radiographic findings of diffuse alveolar filling differ from those in the more chronic forms of silicosis. Histologic findings similar to pulmonary alveolar proteinosis have been described, and extrapulmonary (renal and hepatic) abnormalities are occasionally reported. Rapid progression to severe hypoxaemic ventilatory failure is the usual course.
Tuberculosis may complicate all forms of silicosis, but people with acute and accelerated disease may be at highest risk. Silica exposure alone, even without silicosis may also predispose to this infection. M. tuberculosis is the usual organism, but atypical mycobacteria are also seen.
Even in the absence of radiographic silicosis, silica-exposed workers may also have other diseases associated with occupational dust exposure, such as chronic bronchitis and the associated emphysema. These abnormalities are associated with many occupational mineral dust exposures, including dusts containing silica.
Pathogenesis and the Association with Tuberculosis
The precise pathogenesis of silicosis is uncertain, but an abundance of evidence implicates the interaction between the pulmonary alveolar macrophage and silica particles deposited in the lung. Surface properties of the silica particle appear to promote macrophage activation. These cells then release chemotactic factors and inflammatory mediators that result in a further cellular response by polymorphonuclear leukocytes, lymphocytes and additional macrophages. Fibroblast-stimulating factors are released that promote hyalinization and collagen deposition. The resulting pathologic silicotic lesion is the hyaline nodule, containing a central acellular zone with free silica surrounded by whorls of collagen and fibroblasts, and an active peripheral zone composed of macrophages, fibroblasts, plasma cells, and additional free silica as shown in figure 1.
Figure 1. Typical silicotic nodule, microscopic section. Courtesy of Dr. V. Vallyathan.
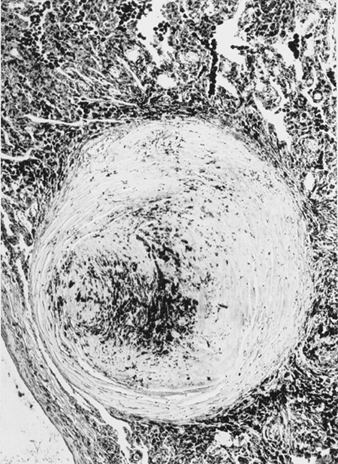
The precise properties of silica particles that evoke the pulmonary response described above are not known, but surface characteristics may be important. The nature and the extent of the biological response are in general related to the intensity of the exposure; however, there is growing evidence that freshly fractured silica may be more toxic than aged dust containing silica, an effect perhaps related to reactive radical groups on the cleavage planes of freshly fractured silica. This may offer a pathogenic explanation for the observation of cases of advanced disease in both sandblasters and rock drillers where exposures to recently fractured silica are particularly intense.
The initiating toxic insult may occur with minimal immunological reaction; however, a sustained immunological response to the insult may be important in some of the chronic manifestations of silicosis. For example, antinuclear antibodies may occur in accelerated silicosis and scleroderma, as well as other collagen diseases in workers who have been exposed to silica. The susceptibility of silicotic workers to infections, such as tuberculosis and Nocardia asteroides, is likely related to the toxic effect of silica on pulmonary macrophages.
The link between silicosis and tuberculosis has been recognized for nearly a century. Active tuberculosis in silicotic workers may exceed 20% when community prevalence of tuberculosis is high. Again, people with acute silicosis appear to be at considerably higher risk.
Clinical Picture of Silicosis
The primary symptom is usually dyspnoea, first noted with activity or exercise and later at rest as the pulmonary reserve of the lung is lost. However, in the absence of other respiratory disease, shortness of breath may be absent and the presentation may be an asymptomatic worker with an abnormal chest radiograph. The radiograph may at times show quite advanced disease with only minimal symptoms. The appearance or progression of dyspnoea may herald the development of complications including tuberculosis, airways obstruction or PMF. Cough is often present secondary to chronic bronchitis from occupational dust exposure, tobacco use, or both. Cough may at times also be attributed to pressure from large masses of silicotic lymph nodes on the trachea or mainstem bronchi.
Other chest symptoms are less common than dyspnoea and cough. Haemoptysis is rare and should raise concern for complicating disorders. Wheeze and chest tightness may occur usually as part of associated obstructive airways disease or bronchitis. Chest pain and finger clubbing are not features of silicosis. Systemic symptoms, such as fever and weight loss, suggest complicating infection or neoplastic disease. Advanced forms of silicosis are associated with progressive respiratory failure with or without cor pulmonale. Few physical signs may be noted unless complications are present.
Radiographic Patterns and Functional Pulmonary Abnormalities
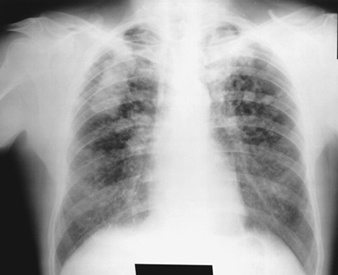
The earliest radiographic signs of uncomplicated silicosis are generally small rounded opacities. These can be described by the ILO International Classification of Radiographs of Pneumoconioses by size, shape and profusion category. In silicosis, “q” and “r” type opacities dominate. Other patterns including linear or irregular shadows have also been described. The opacities seen on the radiograph represent the summation of pathologic silicotic nodules. They are usually found predominantly in the upper zones and may later progress to involve other zones. Hilar lymphadenopathy is also noted sometimes in advance of nodular parenchymal shadows. Egg shell calcification is strongly suggestive of silicosis, although this feature is seen infrequently. PMF is characterized by the formation of large opacities. These large lesions can be described by size using the ILO classification as categories A, B or C. Large opacities or PMF lesions tend to contract, usually to the upper lobes, leaving areas of compensatory emphysema at their margins and often in the lung bases. As a result, previously evident small rounded opacities may disappear at times or be less prominent. Pleural abnormalities may occur but are not a frequent radiographic feature in silicosis. Large opacities may also pose concern regarding neoplasm and radiographic distinction in the absence of old films may be difficult. All lesions that cavitate or change rapidly should be evaluated for active tuberculosis. Acute silicosis may present with a radiologic alveolar filling pattern with rapid development of PMF or complicated mass lesions. See figures 2 and 3.
Figure 2. Chest radiograph, acute silico-proteinosis in a surface coal mine driller. Courtesy of Dr. NL Lapp and Dr. DE Banks.
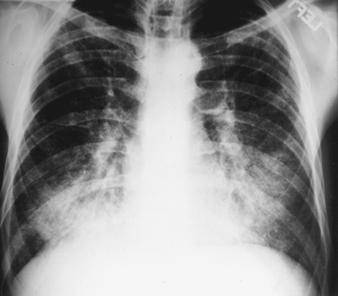
Figure 3. Chest radiograph, complicated silicosis demonstrating progressive massive fibrosis.
Pulmonary function tests, such as spirometry and diffusing capacity, are helpful for the clinical evaluation of people with suspected silicosis. Spirometry may also be of value in early recognition of the health effects from occupational dust exposures, as it may detect physiologic abnormalities that may precede radiologic changes. No solely characteristic pattern of ventilatory impairment is present in silicosis. Spirometry may be normal, or when abnormal, the tracings may show obstruction, restriction or a mixed pattern. Obstruction may indeed be the more common finding. These changes tend to be more marked with advanced radiologic categories. However, poor correlation exists between radiographic abnormalities and ventilatory impairment. In acute and accelerated silicosis, functional changes are more marked and progression is more rapid. In acute silicosis, radiologic progression is accompanied by increasing ventilatory impairment and gas exchange abnormalities, which leads to respiratory failure and eventually to death from intractable hypoxaemia.
Complications and Special Diagnostic Issues
With a history of exposure and a characteristic radiograph, the diagnosis of silicosis is generally not difficult to establish. Challenges arise only when the radiologic features are unusual or the history of exposure is not recognized. Lung biopsy is rarely required to establish the diagnosis. However, tissue samples are helpful in some clinical settings when complications are present or the differential diagnosis includes tuberculosis, neoplasm or PMF. Biopsy material should be sent for culture, and in research settings, dust analysis may be a useful additional measure. When tissue is required, open lung biopsy is generally necessary for adequate material for examination.
Vigilance for infectious complications, especially tuberculosis, cannot be overemphasized, and symptoms of change in cough or hemoptysis, and fever or weight loss should trigger a work-up to exclude this treatable problem.
Substantial concern and interest about the relationship between silica exposure, silicosis and cancer of the lung continues to stimulate debate and further research. In October of 1996, a committere of The International Agency for Research on Cancer (IARC) classified crystalline silica as a Group I carcinogen, reaching this conclusion based on “sufficient evidence of carcinogenicity in humans”. Uncertainty over the pathogenic mechanisms for the development of lung cancer in silica-exposed populations exists, and the possible relationship between silicosis (or lung fibrosis) and cancer in exposed workers continues to be studied. Regardless of the mechanism that may be responsible for neoplastic events, the known association between silica exposures and silicosis dictates controlling and reducing exposures to workers at risk for this disease.
Prevention of Silicosis
Prevention remains the cornerstone of eliminating this occupational lung disease. The use of improved ventilation and local exhaust, process enclosure, wet techniques, personal protection including the proper selection of respirators, and where possible, industrial substitution of agents less hazardous than silica all reduce exposure. The education of workers and employers regarding the hazards of silica dust exposure and measures to control exposure is also important.
If silicosis is recognized in a worker, removal from continuing exposure is advisable. Unfortunately, the disease may progress even without further silica exposure. Additionally, finding a case of silicosis, especially the acute or accelerated form, should prompt a workplace evaluation to protect other workers also at risk.
Screening and Surveillance
Silica and other mineral-dust exposed workers should have periodic screening for adverse health effects as a supplement to, but not a substitute for, dust exposure control. Such screening commonly includes evaluations for respiratory symptoms, lung function abnormalities, and neoplastic disease. Evaluations for tuberculosis infection should also be performed. In addition to individual worker screening, data from groups of workers should be collected for surveillance and prevention activities. Guidance for these types of studies is included in the list of suggested readings.
Therapy, Management of Complications and Control of Silicosis
When prevention has been unsuccessful and silicosis has developed, therapy is directed largely at complications of the disease. Therapeutic measures are similar to those commonly used in the management of airway obstruction, infection, pneumothorax, hypoxaemia, and respiratory failure complicating other pulmonary disease. Historically, the inhalation of aerosolized aluminium has been unsuccessful as a specific therapy for silicosis. Polyvinyl pyridine-N-oxide, a polymer that has protected experiment animals, is not available for use in humans. Recent laboratory work with tetrandrine has shown in vivo reduction in fibrosis and collagen synthesis in silica exposed animals treated with this drug. However, strong evidence of human efficacy is currently lacking, and there are concerns about the potential toxicity, including the mutagenicity, of this drug. Because of the high prevalence of disease in some countries, investigations of combinations of drugs and other interventions continue. Currently, no successful approach has emerged, and the search for a specific therapy for silicosis to date has been unrewarding.
Further exposure is undesirable, and advice on leaving or changing the current job should be given with information about past and present exposure conditions.
In the medical management of silicosis, vigilance for complicating infection, especially tuberculosis, is critical. The use of BCG in the tuberculin-negative silicotic patient is not recommended, but the use of preventive isoniazid (INH) therapy in the tuberculin-positive silicotic subject is advised in countries where the prevalence of tuberculosis is low. The diagnosis of active tuberculosis infection in patients with silicosis can be difficult. Clinical symptoms of weight loss, fever, sweats and malaise should prompt radiographic evaluation and sputum acid-fast bacilli strains and cultures. Radiographic changes, including enlargement or cavitation in conglomerate lesions or nodular opacities, are of particular concern. Bacteriological studies on expectorated sputum may not always be reliable in silicotuberculosis. Fiberoptic bronchoscopy for additional specimens for culture and study may often be helpful in establishing a diagnosis of active disease. The use of multidrug therapy for suspected active disease in silicotics is justified at a lower level of suspicion than in the non-silicotic subject, due to the difficulty in firmly establishing evidence for active infection. Rifampin therapy appears to have enhanced the success rate of treatment of silicosis complicated by tuberculosis, and in some recent studies response to short-term therapy was comparable in cases of silicotuberculosis to that in matched cases of primary tuberculosis.
Ventilatory support for respiratory failure is indicated when precipitated by a treatable complication. Pneumothorax, spontaneous and ventilator-related, is usually treated by chest tube insertion. Bronchopleural fistula may develop, and surgical consultation and management should be considered.
Acute silicosis may rapidly progress to respiratory failure. When this disease resembles pulmonary alveolar proteinosis and severe hypoxaemia is present, aggressive therapy has included massive whole-lung lavage with the patient under general anaesthesia in an attempt to improve gas exchange and remove alveolar debris. Although appealing in concept, the efficacy of whole lung lavage has not been established. Glucocorticoid therapy has also been used for acute silicosis; however, it is still of unproven benefit.
Some young patients with end-stage silicosis may be considered candidates for lung or heart-lung transplantation by centres experienced with this expensive and high-risk procedure. Early referral and evaluation for this intervention may be offered to selected patients.
The discussion of an aggressive and high-technology therapeutic intervention such as transplantation serves dramatically to underscore the serious and potentially fatal nature of silicosis, as well as to emphasize the crucial role for primary prevention. The control of silicosis ultimately depends upon the reduction and control of workplace dust exposures. This is accomplished by rigorous and conscientious application of fundamental occupational hygiene and engineering principles, with a commitment to the preservation of worker health.
Aetiopathogensis of pneumoconioses
Pneumoconioses have been recognized as occupational diseases for a long time. Substantial efforts have been directed to research, primary prevention and medical management. But physicians and hygienists report that the problem is still present in both industrialized and industrializing countries (Valiante, Richards and Kinsley 1992; Markowitz 1992). As there is strong evidence that the three main industrial minerals responsible for the pneumoconioses (asbestos, coal and silica) will continue to have some economical importance, thus further entailing possible exposure, it is expected that the problem will continue to be of some magnitude throughout the world, particularly among underserved populations in small industries and small mining operations. Practical difficulties in primary prevention, or insufficient understanding of the mechanisms responsible for the induction and the progression of the disease are all factors which could possibly explain the continuing presence of the problem.
The aetiopathogenesis of pneumoconioses can be defined as the appraisal and understanding of all the phenomena occurring in the lung following the inhalation of fibrogenic dust particles. The expression cascade of events is often found in the literature on the subject. The cascade is a series of events that first exposure and at its farthest extent progresses to the disease in its more severe forms. If we except the rare forms of accelerated silicosis, which can develop after only a few months of exposure, most of the pneumoconioses develop following exposure periods measured in decades rather than years. This is especially true nowadays in workplaces adopting modern standards of prevention. Aetiopathogenesis phenomena should thus be analysed in terms of its long-term dynamics.
In the last 20 years, a large amount of information has become available on the numerous and complex pulmonary reactions involved in interstitial lung fibrosis induced by several agents, including mineral dusts. These reactions were described at the biochemical and cellular level (Richards, Masek and Brown 1991). Contributions were made by not only physicists and experimental pathologists but also by clinicians who used bronchoalveolar lavage extensively as a new pulmonary technique of investigation. These studies pictured aetiopathogenesis as a very complex entity, which can nonetheless be broken down to reveal several facets: (1) the inhalation itself of dust particles and the consequent constitution and significance of the pulmonary burden (exposure-dose-response relationships), (2) the physicochemical characteristics of the fibrogenic particles, (3) biochemical and cellular reactions inducing the fundamental lesions of the pneumoconioses and (4) the determinants of progression and complication. The later facet must not be ignored, since the more severe forms of pneumoconioses are the ones which entail impairment and disability.
A detailed analysis of the aetiopathogenesis of the pneumoconioses is beyond the scope of this article. One would need to distinguish the several types of dust and to go deeply into numerous specialized areas, some of which are still the subject of active research. But interesting general notions emerge from the currently available amount of knowledge on the subject. They will be presented here through the four “facets” previously mentioned and the bibliography will refer the interested reader to more specialized texts. Examples will be essentially given for the three main and most documented pneumoconioses: asbestosis, coal workers’ pneumoconioses (CWP) and silicosis. Possible impacts on prevention will be discussed.
Exposure-Dose-Response Relationships
Pneumoconioses result from the inhalation of certain fibrogenic dust particles. In the physics of aerosols, the term dust has a very precise meaning (Hinds 1982). It refers to airborne particles obtained by mechanical comminution of a parent material in a solid state. Particles generated by other processes should not be called dust. Dust clouds in various industrial settings (e.g., mining, tunnelling, sand blasting and manufacturing) generally contain a mixture of several types of dust. The airborne dust particles do not have a uniform size. They exhibit a size distribution. Size and other physical parameters (density, shape and surface charge) determine the aerodynamic behaviour of the particles and the probability of their penetration and deposition in the several compartments of the respiratory system.
In the field of pneumoconioses, the site compartment of interest is the alveolar compartment. Airborne particles small enough to reach this compartments are referred to as respirable particles. All particles reaching the alveolar compartments are not systematically deposited, some being still present in the exhaled air. The physical mechanisms responsible for deposition are now well understood for isometric particles (Raabe 1984) as well as for fibrous particles (Sébastien 1991). The functions relating the probability of deposition to the physical parameters have been established. Respirable particles and particles deposited in the alveolar compartment have slightly different size characteristics. For non-fibrous particles, size-selective air sampling instruments and direct reading instruments are used to measure mass concentrations of respirable particles. For fibrous particles, the approach is different. The measuring technique is based upon filter collection of “total dust” and counting of fibres under the optical microscope. In this case, the size selection is made by excluding from the count the “non-respirable” fibres with dimensions exceeding predetermined criteria.
Following the deposition of particles on the alveolar surfaces there starts the so-called alveolar clearance process. Chemotactic recruitment of macrophages and phagocytosis constitute its first phases. Several clearance pathways have been described: removal of dust-laden macrophages toward the ciliated airways, interaction with the epithelial cells and transfer of free particles through the alveolar membrane, phagocytosis by interstitial macrophages, sequestration into the interstitial area and transportation to the lymph nodes (Lauweryns and Baert 1977). Clearance pathways have specific kinetics. Not only the exposure regimen, but also the physicochemical characteristics of the deposited particles, trigger the activation of the different pathways responsible for the lung’s retention of such contaminants.
The notion of a retention pattern specific to each type of dust is rather new, but is now sufficiently established to be integrated into aetiopathogenesis schemes. For example, this author has found that after long term exposure to asbestos, fibres will accumulate in the lung if they are of the amphibole type, but will not if they are of the chrysotile type (Sébastien 1991). Short fibres have been shown to be cleared more rapidly than longer ones. Quartz is known to exhibit some lymph tropism and readily penetrates the lymphatic system. Modifying the surface chemistry of quartz particles has been shown to affect alveolar clearance (Hemenway et al. 1994; Dubois et al. 1988). Concomitant exposure to several dust types may also influence alveolar clearance (Davis, Jones and Miller 1991).
During alveolar clearance, dust particles may undergo some chemical and physical changes. Examples of theses changes include coating with ferruginous material, the leaching of some elemental constituents and the adsorption of some biological molecules.
Another notion recently derived from animal experiments is that of “lung overload” (Mermelstein et al. 1994). Rats heavily exposed by inhalation to a variety of insoluble dusts developed similar responses: chronic inflammation, increased numbers of particle-laden macrophages, increased numbers of particles in the interstitium, septal thickening, lipoproteinosis and fibrosis. These findings were not attributed to the reactivity of the dust tested (titanium dioxide, volcanic ash, fly ash, petroleum coke, polyvinyl chloride, toner, carbon black and diesel exhaust particulates), but to an excessive exposure of the lung. It is not known if lung overload must be considered in the case of human exposure to fibrogenic dusts.
Among the clearance pathways, the transfer towards the interstitium would be of particular importance for pneumoconioses. Clearance of particles having undergone sequestration into the interstitium is much less effective than clearance of particles engulfed by macrophages in the alveolar space and removed by ciliated airways (Vincent and Donaldson 1990). In humans, it was found that after long-term exposure to a variety of inorganic airborne contaminants, the storage was much greater in interstitial than alveolar macrophages (Sébastien et al. 1994). The view was also expressed that silica-induced pulmonary fibrosis involves the reaction of particles with interstitial rather than alveolar macrophages (Bowden, Hedgecock and Adamson 1989). Retention is responsible for the “dose”, a measure of the contact between the dust particles and their biological environment. A proper description of the dose would require that one know at each point in time the amount of dust stored in the several lung structures and cells, the physicochemical states of the particles (including the surface states), and the interactions between the particles and the pulmonary cells and fluids. Direct assessment of dose in humans is obviously an impossible task, even if methods were available to measure dust particles in several biological samples of pulmonary origin such as sputum, bronchoalveolar lavage fluid or tissue taken at biopsy or autopsy (Bignon, Sébastien and Bientz 1979). These methods were used for a variety of purposes: to provide information on retention mechanisms, to validate certain exposure information, to study the role of several dust types in pathogenic developments (e.g., amphiboles versus chrysotile exposure in asbestosis or quartz versus coal in CWP) and to assist in diagnosis.
But these direct measurements provide only a snapshot of retention at the time of sampling and do not allow the investigator to reconstitute dose data. New dosimetric models offer interesting perspectives in that regard (Katsnelson et al. 1994; Smith 1991; Vincent and Donaldson 1990). These models aim at assessing dose from exposure information by considering the probability of deposition and the kinetics of the different clearance pathways. Recently there was introduced into these models the interesting notion of “harmfulness delivery” (Vincent and Donaldson 1990). This notion takes into account the specific reactivity of the stored particles, each particle being considered as a source liberating some toxic entities into the pulmonary milieu. In the case of quartz particles for example, it could be hypothesized that some surface sites could be the source of active oxygen species. Models developed along such lines could also be refined to take into account the great interindividual variation generally observed with alveolar clearance. This was experimentally documented with asbestos, “high retainer animals” being at greater risk of developing asbestosis (Bégin and Sébastien 1989).
So far, these models were exclusively used by experimental pathologists. But they could also be useful to epidemiologists (Smith 1991). Most epidemiological studies looking at exposure response relationships relied on “cumulative exposure”, an exposure index obtained by integrating over time the estimated concentrations of airborne dust to which workers had been exposed (product of intensity and duration). The use of cumulative exposure has some limitations. Analyses based on this index implicitly assume that duration and intensity have equivalent effects on risk (Vacek and McDonald 1991).
Maybe the use of these sophisticated dosimetric models could provide some explanation for a common observation in the epidemiology of pneumoconioses: “the considerable between-work force differences” and this phenomenon was clearly observed for asbestosis (Becklake 1991) and for CWP (Attfield and Morring 1992). When relating the prevalence of the disease to the cumulative exposure, great differences—up to 50-fold—in risk were observed between some occupational groups. The geological origin of the coal (coal rank) provided partial explanation for CWP, mining deposits of high rank coal (a coal with high carbon content, like anthracite) yielding greater risk. The phenomenon remains to be explained in the case of asbestosis. Uncertainties on the proper exposure response curve have some bearings—at least theoretically—on the outcome, even at current exposure standards.
More generally, exposure metrics are essential in the process of risk assessment and the establishment of control limits. The use of the new dosimetric models may improve the process of risk assessment for pneumoconioses with the ultimate goal of increasing the degree of protection offered by control limits (Kriebel 1994).
Physicochemical Characteristics of Fibrogenic Dust Particles
A toxicity specific to each type of dust, related to the physicochemical characteristics of the particles (including the more subtle ones such as the surface characteristics), constitutes probably the most important notion to have emerged progressively during the last 20 years. In the very earliest stages of research, no differentiation were made among “mineral dusts”. Then generic categories were introduced: asbestos, coal, artificial inorganic fibres, phyllosilicates and silica. But this classification was found to be not precise enough to account for the variety in observed biological effects. Nowadays a mineralogical classification is used. For example, the several mineralogical types of asbestos are distinguished: serpentine chrysotile, amphibole amosite, amphibole crocidolite and amphibole tremolite. For silica, a distinction is generally made between quartz (by far the most prevalent), other crystalline polymorphs, and amorphous varieties. In the field of coal, high rank and low rank coals should be treated separately, since there is strong evidence that the risk of CWP and especially the risk of progressive massive fibrosis is much greater after exposure to dust produced in high rank coal mines.
But the mineralogical classification has also some limits. There is evidence, both experimental and epidemiological (taking into account “between-workforce differences”), that the intrinsic toxicity of a single mineralogical type of dust can be modulated by acting on the physicochemical characteristics of the particles. This raised the difficult question of the toxicological significance of each of the numerous parameters which can be used to describe a dust particle and a dust cloud. At the single particle level, several parameters can be considered: bulk chemistry, crystalline structure, shape, density, size, surface area, surface chemistry and surface charge. Dealing with dust clouds adds another level of complexity because of the distribution of these parameters (e.g., size distribution and the composition of mixed dust).
The size of the particles and their surface chemistry were the two parameters most studied to explain the modulation effect. As seen before, retention mechanisms are size related. But size may also modulate the toxicity in situ, as demonstrated by numerous animal and in vitro studies.
In the field of mineral fibres, the size was considered of so much importance that it constituted the basis of a pathogenesis theory. This theory attributed the toxicity of fibrous particles (natural and artificial) to the shape and size of the particles, leaving no role for the chemical composition. In dealing with fibres, size must be broken down into length and diameter. A two-dimensional matrix should be used to report size distributions, the useful ranges being 0.03 to 3.0mm for diameter and 0.3 to 300mm for length (Sébastien 1991). Integrating the results of the numerous studies, Lippman (1988) assigned a toxicity index to several cells of the matrix. There is a general tendency to believe that long and thin fibres are the most dangerous ones. Since the standards currently used in industrial hygiene are based upon the use of the optical microscope, they ignore the thinnest fibres. If assessing the specific toxicity of each cell within the matrix has some academic interest, its practical interest is limited by the fact that each type of fibre is associated with a specific size distribution that is relatively uniform. For compact particles, such as coal and silica, there is unclear evidence about a possible specific role for the different size sub-fractions of the particles deposited in the alveolar region of the lung.
More recent pathogenesis theories in the field of mineral dust imply active chemical sites (or functionalities) present at the surface of the particles. When the particle is “born” by separation from its parent material, some chemical bonds are broken in either a heterolytic or a homolytic way. What occurs during breaking and subsequent recombinations or reactions with ambient air molecules or biological molecules makes up the surface chemistry of the particles. Regarding quartz particles for example, several chemical functionalities of special interest have been described: siloxane bridges, silanol groups, partially ionized groups and silicon-based radicals.
These functionalities can initiate both acid-base and redox reactions. Only recently has attention been drawn to the latter (Dalal, Shi and Vallyathan 1990; Fubini et al. 1990; Pézerat et al. 1989; Kamp et al. 1992; Kennedy et al. 1989; Bronwyn, Razzaboni and Bolsaitis 1990). There is now good evidence that particles with surface-based radicals can produce reactive oxygen species, even in a cellular milieu. It is not certain if all the production of oxygen species should be attributed to the surface-based radicals. It is speculated that these sites may trigger the activation of lung cells (Hemenway et al. 1994). Other sites may be involved in the membranolytic activity of the cytotoxic particles with reactions such as ionic attraction, hydrogen bonding and hydrophobic bonding (Nolan et al. 1981; Heppleston 1991).
Following the recognition of surface chemistry as an important determinant of dust toxicity, several attempts were made to modify the natural surfaces of mineral dust particles to reduce their toxicity, as assessed in experimental models.
Adsorption of aluminium on quartz particles was found to reduce their fibrogenicity and to favour alveolar clearance (Dubois et al. 1988). Treatment with polyvinylpyridine-N-oxide (PVPNO) had also some prophylactic effect (Goldstein and Rendall 1987; Heppleston 1991). Several other modifying processes were used: grinding, thermal treatment, acid etching and adsorption of organic molecules (Wiessner et al. 1990). Freshly fractured quartz particles exhibited the highest surface activity (Kuhn and Demers 1992; Vallyathan et al. 1988). Interestingly enough, every departure from this “fundamental surface” led to a decrease in quartz toxicity (Sébastien 1990). The surface purity of several naturally occurring quartz varieties could be responsible for some observed differences in toxicity (Wallace et al. 1994). Some data support the idea that the amount of uncontaminated quartz surface is an important parameter (Kriegseis, Scharman and Serafin 1987).
The multiplicity of the parameters, together with their distribution in the dust cloud, yields a variety of possible ways to report air concentrations: mass concentration, number concentration, surface area concentration and concentration in various size categories. Thus, numerous indices of exposure can be constructed and the toxicological significance of each has to be assessed. The current standards in occupational hygiene reflect this multiplicity. For asbestos, the standards are based on the numerical concentration of fibrous particles in a certain geometrical size category. For silica and coal, the standards are based on the mass concentration of respirable particles. Some standards have also been developed for exposure to mixtures of particles containing quartz. No standard is based upon surface characteristics.
Biological Mechanisms Inducing the Fundamental Lesions
Pneumoconioses are interstitial fibrous lung diseases, the fibrosis being diffuse or nodular. The fibrotic reaction involves the activation of the lung fibroblast (Goldstein and Fine 1986) and the production and metabolism of the connective tissue components (collagen, elastin and glycosaminoglycans). It is considered to represent a late healing stage after lung injury (Niewoehner and Hoidal 1982). Even if several factors, essentially related to the characteristics of exposure, can modulate the pathological response, it is interesting to note that each type of pneumoconiosis is characterized by what could be called a fundamental lesion. The fibrosing alveolitis around the peripheral airways constitutes the fundamental lesion of asbestos exposure (Bégin et al. 1992). The silicotic nodule is the fundamental lesion of silicosis (Ziskind, Jones and Weil 1976). Simple CWP is composed of dust macules and nodules (Seaton 1983).
The pathogenesis of the pneumoconioses is generally presented as a cascade of events whose sequence runs as follows: alveolar macrophage alveolitis, signalling by inflammatory cell cytokines, oxidative damage, proliferation and activation of fibroblasts and the metabolism of collagen and elastin. Alveolar macrophage alveolitis is a characteristic reaction to retention of fibrosing mineral dust (Rom 1991). The alveolitis is defined by increased numbers of activated alveolar macrophages releasing excessive quantities of mediators including oxidants, chemotaxins, fibroblast growth factors and protease. Chemotaxins attract neutrophils and, together with macrophages, may release oxidants capable of injuring alveolar epithelial cells. Fibroblast growth factors gain access to the interstitium, where they signal fibroblasts to replicate and increase the production of collagen.
The cascade starts at the first encounter of particles deposited in the alveoli. With asbestos for example, the initial lung injury occurs almost immediately after exposure at the alveolar duct bifurcations. After only 1 hour of exposure in animal experiments, there is active uptake of fibres by type I epithelial cells (Brody et al. 1981). Within 48 hours, increased numbers of alveolar macrophages accumulate at sites of deposition. With chronic exposure, this process may lead to peribronchiolar fibrosing alveolitis.
The exact mechanism by which deposited particles produce primary biochemical injury to the alveolar lining, a specific cell, or any of its organelles, is unknown. It may be that extremely rapid and complex biochemical reactions result in free radical formation, lipid peroxidation, or a depletion in some species of vital cell protectant molecule. It has been shown that mineral particles can act as catalytic substrates for hydroxyl and superoxide radical generation (Guilianelli et al. 1993).
At the cellular level, there is slightly more information. After deposition at the alveolar level, the very thin epithelial type I cell is readily damaged (Adamson, Young and Bowden 1988). Macrophages and other inflammatory cells are attracted to the damage site and the inflammatory response is amplified by the release of arachidonic acid metabolites such as prostaglandins and leukotrienes together with exposure of the basement membrane (Holtzman 1991; Kuhn et al. 1990; Engelen et al. 1989). At this stage of primary damage, the lung architecture becomes disorganized, showing an interstitial oedema.
During the chronic inflammatory process, both the surface of the dust particles and the activated inflammatory cells release increased amounts of reactive oxygen species in the lower respiratory tract. The oxidative stress in the lung has some detectable effects on the antioxidant defense system (Heffner and Repine 1989), with expression of antioxidant enzymes like superoxide dismutase, glutathione peroxidases and catalase (Engelen et al. 1990). These factors are located in the lung tissue, the interstitial fluid and the circulating erythrocytes. The profiles of antioxidant enzymes may depend on the type of fibrogenic dust (Janssen et al. 1992). Free radicals are known mediators of tissue injury and disease (Kehrer 1993).
Interstitial fibrosis does result from a repair process. There are numerous theories to explain how the repair process takes place. The macrophage/fibroblast interaction has received the greatest attention. Activated macrophages secrete a network of proinflammatory fibrogenic cytokines: TNF, IL-1, transforming growth factor and platelet-derived growth factor. They also produce fibronectin, a cell surface glycoprotein which acts as a chemical attractant and, under some conditions, as a growth stimulant for mesenchymal cells. Some authors consider that some factors are more important than others. For example, special importance was ascribed to TNF in the pathogenesis of silicosis. In experimental animals, it was shown that collagen deposition after silica instillation in mice was almost completely prevented by anti-TNF antibody (Piguet et al. 1990). The release of platelet-derived growth factor and transforming growth factor was presented as playing an important role in the pathogenesis of asbestosis (Brody 1993).
Unfortunately, many of the macrophage/fibroblast theories tend to ignore the potential balance between the fibrogenic cytokines and their inhibitors (Kelley 1990). In fact, the resulting imbalance between oxidizing and antioxidizing agents, proteases and antiproteases, the arachidonic acid metabolites, elastases and collagenases, as well as the imbalances between the various cytokines and growth factors, would determine the abnormal remodelling of the interstitium component towards the several forms of pneumoconioses (Porcher et al. 1993). In pneumoconioses, the balance is clearly directed towards an overwhelming effect of the damaging cytokine activities.
Because type I cells are incapable of division, after the primary insult, the epithelial barrier is replaced with type II cells (Lesur et al. 1992). There is some indication that if this epithelial repair process is successful and that the regenerating type II cells are not damaged further, the fibrogenesis is not likely to proceed. Under some conditions, the repair by the type II cell is taken to excess, resulting in alveolar proteinosis. This process was clearly demonstrated after silica exposure (Heppleston 1991). To what extent the alterations in epithelial cells influence the fibroblasts is uncertain. Thus, it would seem that fibrogenesis is initiated in areas of extensive epithelial damage, as fibroblasts replicate, then differentiate and produce more collagen, fibronectin and other components of the extracelluar matrix.
There is abundant literature on the biochemistry of the several types of collagen formed in pneumoconioses (Richards, Masek and Brown 1991). The metabolism of such collagen and its stability in the lung are important elements of the fibrogenesis process. The same probably holds for the other components of the damaged connective tissue. The metabolism of collagen and elastin is of particular interest in the healing phase since these proteins are so important to lung structure and function. It has been very nicely shown that alterations in the synthesis of these proteins might determine whether emphysema or fibrosis evolves after lung injury (Niewoehner and Hoidal 1982). In the disease state, mechanisms such as an increase in transglutaminase activity could favour the formation of stable protein masses. In some CWP fibrotic lesions, the protein components account for one-third of the lesion, the rest being dust and calcium phosphate.
Considering only collagen metabolism, several stages of fibrosis are possible, some of which are potentially reversible while others are progressive. There is experimental evidence that unless a critical exposure is exceeded, the early lesions can regress and irreversible fibrosis is an unlikely outcome. In asbestosis for example, several types of lung reactions were described (Bégin, Cantin and Massé 1989): a transient inflammatory reaction without lesion, a low retention reaction with fibrotic scar limited to the distal airways, a high inflammatory reaction sustained by the continuous exposure and the weak clearance of the longest fibres.
It can be concluded from these studies that exposure to fibrotic dust particles is able to trigger several complex biochemical and cellular pathways involved in lung injury and repair. Exposure regimen, physicochemical characteristics of the dust particles, and possibly individual susceptibility factors seem to be the determinants of the fine balance among the several pathways. Physicochemical characteristics will determine the type of the ultimate fundamental lesion. Exposure regimen seems to determine the time course of events. There is some indication that sufficiently low exposure regimens can in most cases limit the lung reaction to non-progressive lesions with no disability or impairment.
Medical surveillance and screening always have been part of the strategies for the prevention of pneumoconioses. In that context, the possibility of detecting some early lesions is advantageous. Increased knowledge of pathogenesis paved the way to the development of several biomarkers (Borm 1994) and to the refinement and use of “non-classical” pulmonary investigation techniques such as the measurement of the clearance rate of deposited 99 technetium diethylenetriamine-penta-acetate (99 Tc-DTPA) to assess pulmonary epithelial integrity (O’Brodovich and Coates 1987), and quantitative gallium-67 lung scan to assess inflammatory activity (Bisson, Lamoureux and Bégin 1987).
Several biomarkers were considered in the field of pneumoconioses: sputum macrophages, serum growth factors, serum type III procollagen peptide, red blood cell antioxidants, fibronectin, leucocyte elastase, neutral metalloendopeptidase and elastin peptides in plasma, volatile hydrocarbons in exhaled air and TNF release by peripheral blood monocytes. Biomarkers are conceptually quite interesting, but many more studies are necessary to assess their significance precisely. This validation effort will be quite demanding, since it will require investigators to conduct prospective epidemiological studies. Such an effort was carried out recently for TNF release by peripheral blood monocytes in CWP. TNF was found to be an interesting marker of CWP progression (Borm 1994). Besides the scientific aspects of the significance of biomarkers in the pathogenesis of pneumoconioses, other issues related to the use of biomarkers must be examined carefully (Schulte 1993), namely, opportunities for prevention, impact on occupational medicine and ethical and legal problems.
Progression and Complication of Pneumoconioses
In the early decades of this century, pneumoconiosis was regarded as a disease that disabled the young and killed prematurely. In industrialized countries, it is now generally regarded as no more than a radiological abnormality, without impairment or disability (Sadoul 1983). However, two observations should be set against this optimistic statement. First, even if under limited exposure, pneumoconiosis remains a relatively silent and asymptomatic disease, it should be known that the disease may progress towards more severe and disabling forms. Factors affecting this progression are definitely important to consider as part of the aetiopathogenesis of the condition. Secondly, there is now evidence that some pneumoconioses can affect general health outcome and can be a contributing factor for lung cancer.
The chronic and progressive nature of asbestosis has been documented from the initial subclinical lesion to clinical asbestosis (Bégin, Cantin and Massé 1989). Modern pulmonary investigation techniques (BAL, CT scan, gallium-67 lung uptake) revealed that inflammation and injury was continuous from the time of exposure, through the latent or subclinical phase, to the development of the clinical disease. It has been reported (Bégin et al. 1985) that 75% of subjects who initially had a positive gallium-67 scan but did not have clinical asbestosis at that time, did progress to “full-blown” clinical asbestosis over a four-year period. In both humans and experimental animals, asbestosis may progress after disease recognition and exposure cessation. It is highly probable that exposure history prior to recognition is an important determinant of progression. Some experimental data support the notion of non-progressive asbestosis associated with light induction exposure and exposure cessation at recognition (Sébastien, Dufresne and Bégin 1994). Assuming that the same notion applies to humans, it would be of the first importance to establish precisely the metrics of “light induction exposure”. In spite of all the efforts at screening working populations exposed to asbestos, this information is still lacking.
It is well-known that asbestos exposure can yield to an excessive risk of lung cancer. Even if it is admitted that asbestos is a carcinogen per se, it has long been debated whether the risk of lung cancer among asbestos workers was related to the exposure to asbestos or to the lung fibrosis (Hughes and Weil 1991). This issue is not resolved yet.
Owing to continuous improvement of working conditions in modern mining facilities, nowadays, CWP is a disease affecting essentially retired miners. If simple CWP is a condition without symptoms and without demonstrable effect on lung function, progressive massive fibrosis (PMF) is a much more severe condition, with major structural alterations of the lung, deficits of lung function and reduced life expectancy. Many studies have aimed at identifying the determinants of progression towards PMF (heavy retention of dust in the lung, coal rank, mycobacterial infection or immunological stimulation). A unifying theory was proposed (Vanhee et al. 1994), based upon a continuous and severe alveolar inflammation with activation of the alveolar macrophages and substantial production of reactive oxygen species, chemotactic factors and fibronectin. Other complications of CWP include mycobacterial infection, Caplan’s syndrome and scleroderma. There is no evidence of elevated risk of lung cancer among coal miners.
The chronic form of silicosis follows exposure, measured in decades rather than years, to respirable dust containing generally less than 30% quartz. But in case of uncontrolled exposure to quartz-rich dust (historical exposures with sand blasting, for example), acute and accelerated forms can be found after only several months. Cases of acute and accelerated disease are particularly at risk of complication by tuberculosis (Ziskind, Jones and Weil 1976). Progression may also occur, with the development of large lesions that obliterate lung structure, called either complicated silicosis or PMF.
A few studies examined the progression of silicosis in relation to exposure and yielded diverging results about the relationships between progression and exposure, before and after onset (Hessel et al. 1988). Recently, Infante-Rivard et al. (1991) studied the prognostic factors influencing the survival of compensated silicotic patients. Patients with small opacities alone on their chest radiograph and who did not have dyspnoea, expectoration or abnormal breath sounds had a survival similar to that of the referents. Other patients had a poorer survival. Finally, one should mention the recent concern about silica, silicosis and lung cancer. There is some evidence for and against the proposition that silica per se is carcinogenic (Agius 1992). Silica may synergize potent environmental carcinogens, such as those in tobacco smoke, through a relatively weak promoting effect on carcinogenesis or by impairing their clearance. Moreover, the disease process associated with or leading to silicosis might carry an increased risk of lung cancer.
Nowadays, progression and complication of pneumoconioses could be considered as a key issue for medical management. The use of classical pulmonary investigation techniques has been refined for early recognition of the disease (Bégin et al. 1992), at a stage where pneumoconiosis is limited to its radiological manifestation, without impairment or disability. In the near future, it is probable that a battery of biomarkers will be available to document even earlier stages of the disease. The question of whether a worker diagnosed with pneumoconiosis—or documented to be in its earlier stages—should be allowed to continue with his or her job has puzzled occupational health decision makers for some time. It is a rather difficult question which entails ethical, social and scientific considerations. If an overwhelming scientific literature is available on the induction of pneumoconiosis, the information on progression usable by decision makers is rather sparse and somewhat confusing. A few attempts were made to study the roles of variables such as exposure history, dust retention and medical condition at onset. The relationships between all these variables do complicate the issue. Recommendations are made for health screening and surveillance of workers exposed to mineral dust (Wagner 1996). Programmes are already—or will be—put in place accordingly. Such programmes would definitely benefit from better scientific knowledge on progression, and especially on the relation between exposure and retention characteristics.
Discussion
The information brought by many scientific disciplines to bear upon the aetiopathogenesis of the pneumoconioses is overwhelming. The major difficulty now is to reassemble the scattered elements of the puzzle into unifying mechanistic pathways leading to the fundamental lesions of the pneumoconioses. Without this necessary integration, we would be left with the contrast between a few fundamental lesions, and very numerous biochemical and cellular reactions.
Our knowledge of aetiopathogenesis has so far influenced the practices of occupational hygiene only to a limited extent, in spite of the strong intention of hygienists to operate according to standards having some biological significance. Two main notions were incorporated in their practices: the size selection of respirable dust particles and the dust type dependence of toxicity. The latter yielded some limits specific to each type of dust. The quantitative risk assessment, a necessary step in defining exposure limits, constitutes a complicated exercise for several reasons, such as the variety of possible exposure indices, poor information on past exposure, the difficulty one has with epidemiological models in dealing with multiple indices of exposure and the difficulty in estimating dose from exposure information. The current exposure limits, embodying sometimes considerable uncertainty, are probably low enough to offer good protection. The between-workforce differences observed in exposure-response relationships however, reflect our incomplete control of the phenomenon.
The impact of newer understanding of the cascade of events in the pathogenesis of the pneumoconioses has not modified the traditional approach to workers’ surveillance, but has significantly helped physicians in their capacity of recognizing the disease (pneumoconiosis) early, at a time when the disease has had only a limited impact on lung function. It is indeed subjects at the early stage of disease that should be recognized and withdrawn from further significant exposure if prevention of disability is to be achieved by medical surveillance.
ILO International Classification of Radiographs of Pneumoconioses
Despite all the national and international energies devoted to their prevention, pneumoconioses are still very present both in industrialized and developing countries, and are responsible for the disability and impairment of many workers. This is why the International Labour Office (ILO), the World Health Organization (WHO) and many national institutes for occupational health and safety continue their fight against these diseases and to propose sustainable programmes for preventing them. For instance, the ILO, the WHO and the US National Institute for Occupational Safety and Health (NIOSH) have proposed in their programmes to work in cooperation on a global fight against silicosis. Part of this programme is based on medical surveillance which includes the reading of thoracic radiographs to help diagnose this pneumoconiosis. This is one example which explains why the ILO, in cooperation with many experts, has developed and updated on a continuous basis a classification of radiographs of pneumoconioses that provides a means for recording systematically the radiographic abnormalities in the chest provoked by the inhalation of dust. The scheme is designed for classifying the appearances of posterio-anterior chest radiographs.
The object of the classification is to codify the radiographic abnormalities of pneumoconioses in a simple, reproducible manner. The classification does not define pathological entities, nor take into account working capacity. The classification does not imply legal definitions of pneumoconioses for compensation purposes, nor imply a level at which compensation is payable. Nevertheless, the classification has been found to have wider uses than anticipated. It is now extensively used internationally for epidemiological research, for the surveillance of those industry occupations and for clinical purposes. Use of the scheme may lead to better international comparability of pneumoconioses statistics. It is also used for describing and recording, in a systematic way, part of the information needed for assessing compensation.
The most important condition for using this system of classification with full value from a scientific and ethical point of view is to read, at all times, films to be classified by systematically referring to the 22 standard films provided in the ILO International Classification set of standard films. If the reader attempts to classify a film without referring to any of the standard films, then no mention of reading according to the ILO International Classification of Radiographs should be made. The possibility of deviating from the classification by over or under reading is so risky that his or her reading should not be used at least for epidemiological research or international comparability of pneumoconioses statistics.
The first classification was proposed for silicosis at the First International Conference of Experts on Pneumoconioses, held in Johannesburg in 1930. It combined both radiographic appearances and impairment of lung functions. In 1958, a new classification based purely on radiographic changes was established (Geneva classification 1958). Since, it has been revised several times, the last time in 1980, always with the objective of providing improved versions to be extensively used for clinical and epidemiological purposes. Each new version of the classification promoted by the ILO has brought modifications and changes based on international experience gained in the use of earlier classifications.
In order to provide clear instructions for the use of the classification, the ILO issued in 1970 a publication entitled International Classification of Radiographs of Pneumoconioses/1968 in the Occupational Safety and Health Series (No. 22). This publication was revised in 1972 as ILO U/C International Classification of Radiographs of Pneumoconioses/1971 and again in 1980 as Guidelines for the use of ILO International Classification of Radiographs of Pneumoconioses, revised edition 1980. The description of standard radiographs is given in table 1.
Table 1. Description of standard radiographs
| 1980 Standard radiographs showing | Small opacities | Pleural thickening | ||||||||||
| Chest wall | ||||||||||||
| Technical quality | Profusion | Shape- size | Extent | Large opacities | Circum- scribed (plaques) | Diffuse | Diaphragm | Costo- phrenic angle obliteration | Pleural calcification | Symbols | Comments | |
| 0/0 (example 1) | 1 | 0/0 | – | – | No | No | No | No | No | No | None | Vascular pattern is well illustrated |
| 0/0 (example 2) | 1 | 0/0 | – | – | No | No | No | No | No | No | None | Also shows vascular pattern, but not as clearly as example 1 |
| 1/1; p/p | 1 | 1/1 | p/p | R L x x x x x x | A | No | No | No | No | No | rp. | Rheumatoid pneumoconiosis in left lower zone. Small opacities are present in all zones, but the profusion in the right-upper zone is typical of (some would say a little more profuse than) that classifiable as category 1/1 |
| 2/2; p/p | 2 | 2/2 | p/p | R L x x x x x x | No | No | No | No | No | No | pi; tb. | Quality defect: radiograph is too light |
| 3/3; p/p | 1 | 3/3 | p/p | R L x x x x x x | No | No | No | No | Yes R L x – | No | ax. | None |
| 1/1; q/q | 1 | 1/1 | q/q | R L x x x x – – | No | No | No | No | No | No | None | Illustrates profusion 1/1 better than shape or size |
| 2/2; q/q | 1 | 2/2 | q/q | R L x x x x x x | No | No | Yes R L x x width: a a extent: 1 1 | No | Yes R L x x | No | None | None |
| 3/3; q/q | 2 | 3/3 | q/q | R L x x x x x x | No | No | No | No | No | No | pi. | Quality defects: poor definition of pleura and cut basal angles |
| 1/1; r/r | 2 | 1/1 | r/r | R L x x x x – – | No | No | No | No | Yes R L – x | No | None | Quality defect: subject movement. Profusion of small opacities is more marked in right lung |
| 2/2; r/r | 2 | 2/2 | r/r | R L x x x x x x | No | No | No | No | No | No | None | Quality defects: radiograph too light and contrast too high. The heart shadow is slightly displaced to the left |
| 3/3; r/r | 1 | 3/3 | r/r | R L x x x x x x | No | No | No | No | No | No | ax; ih. | None |
| 1/1; s/t | 2 | 1/1 | s/t | R L x – x x x x | No | No | No | No | No | No | kl. | Quality defect: cut bases. Kerley lines in lower right zone |
| 2/2; s/s | 2 | 2/2 | s/s | R L – – x x x x | No | No | No | No | No | No | em. | Quality defect: distortion of bases due to shrinking. Emphysema in upper zones |
| 3/3; s/s | 2 | 3/3 | s/s | R L x x x x x x | No | No | Yes R L x x width: a a extent: 3 3 | No | No | No | ho; ih; pi. | Quality defect: radiograph is too light. Honeycomb lung appearance is not marked |
| 1/1; t/t Costophrenic angle obliteration | 1 | 1/1 | t/t | R L – – x x x x | No | No | Yes R L x x width: a a extent: 2 2 | No | Yes R L x – | Yes R L – x extent: 2 | None | This radiograph defines the lower limit for costophrenic angle obliteration. Note shrinkage in lower lung fields |
| 2/2; t/t | 1 | 2/2 | t/t | R L x x x x x x | No | No | Yes R L x x width: a a extent: 1 1 | No | No | No | ih. | Pleural thickening is present in the apices of the lung |
| 3/3; t/t | 1 | 3/3 | t/t | R L x x x x x x | No | No | No | No | No | No | hi; ho; id; ih; tb. | None |
| 1/1; u/u 2/2; u/u 3/3; u/u | – | – | – | – | – | – | – | – | – | – | – | This composite radiograph illustrates the mid-categories of profusion of small opacities classifiable for shape and size as u/u. |
| A | 2 | 2/2 | p/q | R L x x x x x x | A | No | No | No | No | No | No | Quality defects: radiograph is too light and pleural definition is poor |
| B | 1 | 1/2 | p/q | R L x x x x x x | B | No | No | No | No | No | ax; co. | Definition of pleura is slightly imperfect |
| C | 1 | 2/1 | q/t | R L x x x x x x | C | No | No | No | No | No | bu; di; em; es; hi; ih. | The small opacities are difficult to classify because of the presence of the large opacities. Note the left costophrenic angle obliteration. This is not classifiable because it does not reach the lower limit defined by the standard radiograph 1/1; t/t |
| Pleural thickening (circumscribed) | – | – | – | – | – | Yes | No | No | No | No | The pleural thickening present face on, is of indeterminate width, and extent 2 | |
| Pleural thickening (diffuse) | – | – | – | – | – | No | Yes | No | No | Yes | The pleural thickening present in profile, is of width a, and extent 2. Not associated small calcifications | |
| Pleural thickening (calcification) diaphragm | – | – | – | – | – | No | No | Yes | No | Yes | Circumscribed, calcified pleural thickening of extent 2 | |
| Pleural thickening (calcification) chest wall | – | – | – | – | – | Yes | No | No | No | Yes | Calcified and uncalcified pleural thickening present face on, is of indeterminate width, and extent 2 | |
ILO 1980 Classification
The 1980 revision was carried out by the ILO with the cooperation of the Commission of the European Communities, NIOSH and the American College of Radiology. The summary of the classification is given in table 2. It retained the principle of former classifications (1968 and 1971).
Table 2. ILO 1980 International Classification of Radiographs of Pneumoconioses: Summary of details of classification
| Features | Codes | Definitions | |
| Technical quality | |||
| 1 | Good. | ||
| 2 | Acceptable, with no technical defect likely to impair classification of the radiograph of pneumoconiosis. | ||
| 3 | Poor, with some technical defect but still acceptable for classification purposes. | ||
| 4 | Unacceptable. | ||
| Parenchymal abnormalities | |||
| Small opacities | Profusion | The category of profusion is based on assessment of the concentration of opacities by comparison with the standard radiographs. | |
| 0/- 0/0 0/1 1/0 1/1 1/2 2/1 2/2 2/3 3/2 3/3 3/+ | Category O—small opacities absent or less profuse than the lower limit of category 1. Categories 1, 2 and 3—increasing profusion of small opacities as defined by the corresponding standard radiographs. | ||
| Extent | RU RM RL LU LM LL | The zones in which the opacities are seen are recorded. The right (R) and left (L) thorax are both divided into three zones—upper (U), middle (M) and lower (L). The category of profusion is determined by considering the profusion as a whole over the affected zones of the lung and by comparing this with the standard radiographs. | |
| Shape and Size | |||
| Rounded | p/p q/q r/r | The letters p, q and r denote the presence of small, rounded opacities. Three sizes are defined by the appearances on standard radiographs: p = diameter up to about 1.5 mm q = diameter exceeding about 1.5 mm and up to about 3 mm r = diameter exceeding about 3 mm and up to about 10 mm | |
| Irregular | s/s t/t u/u | The letters s, t and u denote the presence of small, irregular opacities. Three sizes are defined by the appearances on standard radiographs: s = width up to about 1.5 mm t = width exceeding about 1.5 mm and up to about 3 mm u = width exceeding 3 mm and up to about 10 mm | |
| Mixed | p/s p/t p/u p/q p/r q/s q/t q/u q/p q/r r/s r/t r/u r/p r/q s/p s/q s/r s/t s/u t/p t/q t/r t/s t/u u/p u/q u/r u/s u/t | For mixed shapes (or sizes) of small opacities, the predominant shape and size is recorded first. The presence of a significant number of another shape and size is recorded after the oblique stroke. | |
| Large opacities | A B C | The categories are defined in terms of the dimensions of the opacities. Category A – an opacity having a greatest diameter exceeding about 10 mm and up to and including 50 mm, or several opacities each greater than about 10 mm, the sum of whose greatest diameters does not exceed about 50 mm. Category B – one or more opacities larger or more numerous than those in category A whose combined area does not exceed the equivalent of the right upper zone. Category C – one or more opacities whose combined area exceeds the equivalent of the right upper zone. | |
| Pleural abnormalities | |||
| Pleural thickening | |||
| Chest wall | Type | Two types of pleural thickening of the chest wall are recognized: circumscribed (plaques) and diffuse. Both types may occur together | |
| Site | R L | Pleural thickening of the chest wall is recorded separately for the right (R) and left (L) thorax. | |
| Width | a b c | For pleural thickening seen along the lateral chest wall the measurement of maximum width is made from the inner line of the chest wall to the inner margin of the shadow seen most sharply at the parenchymal-pleural boundary. The maximum width usually occurs at the inner margin of the rib shadow at its outermost point. a = maximum width up to abut 5 mm b = maximum width over about 5 mm and up to about 10 mm c = maximum width over about 10 mm | |
| Face on | Y N | The presence of pleural thickening seen face-on is recorded even if it can be seen also in profile. If pleural thickening is seen face-on only, width cannot usually be measured. | |
| Extent | 1 2 3 | Extent of pleural thickening is defined in terms of the maximum length of pleural involvement, or as the sum of maximum lengths, whether seen in profile or face-on. 1 = total length equivalent up to one quarter of the projection of the lateral chest wall 2 = total length exceeding one quarter but not one half of the projection of the lateral chest wall 3 = total length exceeding one half of the projection of the lateral chest wall | |
| Diaphragm | Presence | Y N | A plaque involving the diaphragmatic pleura is recorded as present (Y) or absent (N), separately for the right (R) and left (L) thorax. |
| Site | R L | ||
| Costrophrenic angle obliteration | Presence | Y N | The presence (Y) or absence (N) of costophrenic angle obliteration is recorded separately from thickening over other areas, for the right (R) and left (L) thorax. The lower limit for this obliteration is defined by a standard radiograph |
| Site | R L | If the thickening extends up the chest wall, then both costophrenic angle obliteration and pleural thickening should be recorded. | |
| Pleural calcification | Site | The site and extent of pleural calcification are recorded separately for the two lungs, and the extent defined in terms of dimensions. | |
| Chest wall | R L | ||
| Diaphragm | R L | ||
| Other | R L | “Other” includes calcification of the mediastinal and pericardial pleura. | |
| Extent | 1 2 3 | 1 = an area of calcified pleura with greatest diameter up to about 20 mm, or a number of such areas the sum of whose greatest diameters does not exceed about 20 mm. 2 = an area of calcified pleura with greatest diameter exceeding about 20 mm and up to about 100 mm, or a number of such areas the sum of whose greatest diameters exceeds about 20 mm but does not exceed about 100 mm. 3 = an area of calcified pleura with greatest diameter exceeding about 100 mm, or a number of such areas whose sum of greatest diameters exceeds about 100 mm. | |
| Symbols | |||
| It is to be taken that the definition of each of the symbols is preceded by an appropriate word or phrase such as “suspect”, “changes suggestive of”, or “opacities suggestive of”, etc. | |||
| ax | Coalescence of small pneumoconiotic opacities | ||
| bu | Bulla(e) | ||
| ca | Cancer of lung or pleura | ||
| cn | Calcification in small pneumoconiotic opacities | ||
| co | Abnormality of cardiac size or shape | ||
| cp | Cor pulmonale | ||
| cv | Cavity | ||
| di | Marked distortion of the intrathoracic organs | ||
| ef | Effusion | ||
| em | Definite emphysema | ||
| es | Eggshell calcification of hilar or mediastinal lymph nodes | ||
| fr | Fractured rib(s) | ||
| hi | Enlargement of hilar or mediastinal lymph nodes | ||
| ho | Honeycomb lung | ||
| id | Ill-defined diaphragm | ||
| ih | Ill-defined heart outline | ||
| kl | Septal (Kerley) lines | ||
| od | Other significant abnormality | ||
| pi | Pleural thickening in the interlobar fissure of mediastinum | ||
| px | Pneumothorax | ||
| rp | Rheumatoid pneumoconiosis | ||
| tb | Tuberculosis | ||
| Comments | |||
| Presence | Y N | Comments should be recorded pertaining to the classification of the radiograph, particularly if some other cause is thought to be responsible for a shadow which could be thought by others to have been due to pneumoconiosis; also to identify radiographs for which the technical quality may have affected the reading materially. | |
The Classification is based on a set of standard radiographs, a written text and a set of notes (OHS No. 22). There are no features to be seen in a chest radiograph which are pathognomonic of dust exposure. The essential principle is that all appearances which are consistent with those defined and represented in the standard radiographs and the guideline for the use of the ILO International Classification, are to be classified. If the reader believes that any appearance is probably or definitively not dust related, the radiograph should not be classified but an appropriate comment must be added. The 22 standard radiographs have been selected after international trials, in such a way as to illustrate the mid-categories standards of profusion of small opacities and to give examples of category A, B and C standards for large opacities. Pleural abnormalities (diffuse pleural thickening, plaques and obliteration of costophrenic angle) are also illustrated on different radiographs.
Discussion in particular at the Seventh International Pneumoconioses Conference, held in Pittsburgh in 1988, indicated the need for improvement of some parts of the classification, in particular those concerning pleural changes. A discussion group meeting on the revision of the ILO International Classification of Radiographs of Pneumoconioses was convened in Geneva by the ILO in November 1989. The experts made the suggestion that the short classification is of no advantage and can be deleted. As regards pleural abnormalities, the group agreed that this classification would now be divided into three parts: “Diffuse pleural thickening”; “Pleural plaques”; and “Costophrenic angle obliteration”. Diffuse pleural thickening might be divided into chest wall and diaphragm. They were identified according to the six zones—upper, middle and lower, of both right and left lungs. If a pleural thickening is circumscribed, it could be identified as a plaque. All plaques should be measured in centimetres. The obliteration of the costophrenic angle should be systematically noted (whether it exists or not). It is important to identify whether the costophrenic angle is visible or not. This is because of its special importance in relation to pleural diffuse thickening. Whether plaques are classified or not should be merely indicated by a symbol. The flattening of the diaphragm should be recorded by an additional symbol since it is a very important feature in asbestos exposure. The presence of plaques should be recorded in these boxes using the appropriate symbol “c” (calcified) or “h” (hyaline).
A full description of the classification, including its applications and limitation is found in the publication (ILO 1980). The revision of the classification of radiographs is a continuous ILO process, and a revised guideline should be published in the near future (1997-98) taking into account the recommendations of these experts.
Pneumoconioses: Definition
The expression pneumoconiosis, from the Greek pneuma (air, wind) and konis (dust) was coined in Germany by Zenker in 1867 to denote changes in the lungs caused by the retention of inhaled dust. Gradually, the need for distinction between the effects of various types of dust became evident. It was necessary to discriminate among mineral or vegetable dust and their microbiological component. Consequently, the Third International Conference of Experts on Pneumoconiosis, organized by the ILO in Sydney in 1950, adopted the following definition: “Pneumoconiosis is a diagnosable disease of the lungs produced by the inhalation of dust, the term ‘dust’ being understood to refer to particulate matter in the solid phase, but excluding living organisms.”
However, the word disease seems to imply some degree of health impairment which may not be the case with pneumoconioses not connected with the development of lung fibrosis/scarring. In general, the reaction of lung tissue to the presence of dust varies with different dusts. Non-fibrogenic dusts evoke a tissue reaction in lungs characterized by minimal fibrotic reaction and absence of lung function impairment. Such dusts, examples of which are finely divided dusts of kaolinite, titanium dioxide, stannous oxide, barium sulphate and ferric oxide, are frequently referred to as biologically inert.
Fibrogenic dust such as silica or asbestos causes a more pronounced fibrogenic reaction resulting in scars in the lung tissue and obvious disease. The division of dusts into fibrogenic and non-fibrogenic varieties is by no means sharp because there are many minerals, notably silicates, which are intermediate in their ability to produce fibrotic lesions in the lungs. Nevertheless, it proved useful for clinical purposes and is reflected in the classification of pneumoconioses.
A new definition of pneumoconioses was adopted at the Fourth International Conference on Pneumoconiosis, Bucharest, 1971: “Pneumoconiosis is the accumulation of dust in the lungs and the tissue reactions to its presence. For the purpose of this definition, ‘dust’ is meant to be an aerosol composed of solid inanimate particles.”
In order to avoid any misinterpretation, the expression non-neoplastic is sometimes added to the words “tissue reaction”.
The Working Group at the Conference made the following comprehensive statement:
The Definition of Pneumoconiosis
Earlier on, in 1950, a definition of pneumoconiosis was established at the 3rd International Conference of Experts on Pneumoconiosis and this has continued to be used until the present time. In the meantime, the development of new technologies has resulted in more occupational risks, particularly those related to the inhalation of airborne contaminants. Increased knowledge in the field of occupational medicine has enabled new pulmonary diseases of occupational origin to be recognized but has also demonstrated the necessity for a re-examination of the definition of pneumoconiosis established in 1950. The ILO therefore arranged for a Working Group to be convened within the framework of the IVth International Pneumoconiosis Conference in order to examine the question of the definition of pneumoconiosis. The Working Group held a general discussion on the matter and proceeded to examine a number of proposals submitted by its members. It finally adopted a new definition of pneumoconiosis which was prepared together with a commentary. This text is reproduced below.
In recent years a number of countries have included under pneumoconiosis, because of socio-economic reasons, conditions which are manifestly not pneumoconiosis, but are nevertheless occupational pulmonary diseases. Under the term “disease” are included for preventive reasons the earliest manifestations which are not necessarily disabling or life shortening. Therefore the Working Group has undertaken to redefine pneumoconiosis as the accumulation of dust in the lungs and the tissue reactions to its presence. For the purpose of this definition, “dust” is meant to be an aerosol composed of solid inanimate particles. From a pathological point of view pneumoconiosis may be divided for the sake of convenience into collagenous or non-collagenous forms. A non-collagenous pneumoconiosis is caused by a non-fibrogenic dust and has the following characteristics:
- the alveolar architecture remains intact
- the stromal reaction is minimal and consists mainly of reticulin fibres
- the dust reaction is potentially reversible.
Examples of non-collagenous pneumoconiosis are those caused by pure dusts of tin oxide (stannosis) and barium sulphate (barytosis).
Collagenous pneumoconiosis is characterised by:
- permanent alteration or destruction of alveolar architecture
- collagenous stromal reaction of moderate to maximal degree, and
- permanent scarring of lung.
Such collagenous pneumoconiosis may be caused by fibrogenic dusts or by an altered tissue response to a non-fibrogenic dust.
Examples of collagenous pneumoconiosis caused by fibrogenic dusts are silicosis and asbestosis, whereas complicated coalworkers’ pneumoconiosis or progressive massive fibrosis (PMF) is an altered tissue response to a relatively non-fibrogenic dust. In practice, the distinction between collagenous and non-collagenous pneumoconiosis is difficult to establish. Continued exposure to the same dust, such as coal dust, may cause transition from a non-collagenous to a collagenous form. Furthermore, exposure to a single dust is now becoming less common and exposures to mixed dusts having different degrees of fibrogenic potential may result in pneumoconiosis which can range from the non-collagenous to the collagenous forms. There are in addition occupational chronic pulmonary diseases which, although they develop from the inhalation of dust are excluded from the pneumoconiosis because the particles are not known to accumulate in the lungs. The following are examples of potentially disabling occupational chronic pulmonary diseases: byssinosis, berylliosis, farmers’ lung, and related diseases. They have one common denominator, namely the aetiologic component of dust has sensitized the pulmonary or bronchial tissue so that if the lung tissue responds, the inflammation tends to be granulomatous and if the bronchial tissue responds, there is apt to be bronchial constriction. Exposures to noxious inhaled materials in certain industries are associated with an increased risk of mortality from carcinoma of the respiratory tract. Examples of such materials are radioactive ores, asbestos and chromates.
Adopted at the IVth ILO International Conference on Pneumoconiosis. Bucharest, 1971.
Beryllium Disease
Beryllium disease is a systemic disorder involving multiple organs, with pulmonary manifestations being most prominent and common. It occurs on exposure to beryllium in its alloy form or in one of its various chemical compounds. Route of exposure is by inhalation and the disease can be either acute or chronic. Acute disease is extremely rare currently, and none has been reported since the first widespread industrial use of beryllium in the 1940s after industrial hygiene measures had been implemented to limit high-dose exposures. Chronic beryllium disease continues to be reported.
Beryllium, Alloys and Compounds
Beryllium, an industrial substance suspected of having carcinogenic potential, is notable for its lightness in weight, high tensile strength and corrosion resistance. Table 1 outlines the properties of beryllium and its compounds.
Table 1. Properties of beryllium and its compounds
|
Formula |
Specific |
Melting/boiling point (ºC) |
Solubility |
Description |
|
|
Beryllium (Be) |
9.01 (a.w.) |
1.85 |
1,298±5/2,970 |
— |
Grey to silver metal |
|
Beryllium oxide (BeO) |
25 |
3.02 |
2,530±30/— |
Soluble in acids and alkalis; insoluble in water |
White amorphous powder |
|
Beryllium fluoride1 (BeF2 ) |
47.02 |
1.99 |
Sublimes 800 °C |
Readily soluble in water; sparingly soluble in ethyl alcohol |
Hygroscopic solid |
|
Beryllium chloride2 (BeCl2 ) |
79.9 |
1.90 |
405/520 |
Very soluble in water; soluble in ethyl alcohol, benzene, ethyl ether and carbon disulphide |
White or slightly yellow deliquescent crystals |
|
Beryllium nitrate3 (Be(NO3 )2 ·3H2 O) |
187.08 |
1.56 |
60/142 |
Soluble in water and ethyl alcohol |
White to faintly yellow deliquescent crystals |
|
Beryllium nitride4 (Be3 N2 ) |
55.06 |
— |
2,200±100/— |
— |
Hard, refractory white crystals |
|
Beryllium sulphate |
177.2 |
1.71 |
100/— |
Soluble in water; insoluble in ethyl alcohol |
Colourless crystals |
1 Beryllium fluoride is made by the decompensation at 900–950 ºC of ammonium beryllium fluoride. Its main use is in the production of beryllium metal by reduction with magnesium.
2 Beryllium chloride is manufactured by passing chlorine over a mixture of beryllium oxide and carbon.
3 Beryllium nitrate is produced by the action of nitric acid on beryllium oxide. It is used as a chemical reagent and as a gas mantle hardener.
4 Beryllium nitride is prepared by heating beryllium metal powder in an oxygen-free, nitrogen atmosphere at 700–1,400 ºC. It is used in atomic energy reactions, including the production of the radioactive carbon isotope carbon-14.
5 Beryllium sulphate hydrate is produced by treating the fritted ore with concentrated suphuric acid.It is used in the production of metallic beryllium by the sulphate process.
Sources
Beryl (3BeO·Al2O3·6SiO2) is the chief commercial source of beryllium, the most abundant of the minerals containing high concentrations of beryllium oxide (10 to 13%). Major sources of beryl are to be found in Argentina, Brazil, India, Zimbabwe and the Republic of South Africa. In the United States, beryl is found in Colorado, South Dakota, New Mexico and Utah. Bertrandite, a low-grade ore (0.1 to 3%) with an acid-soluble beryllium content, is now being mined and processed in Utah.
Production
The two most important methods of extracting beryllium from the ore are the sulphate process and the fluoride process.
In the sulphate process, crushed beryl is melted in an arc furnace at 1,65°C and poured through a high-velocity water stream to form a frit. After heat treatment, the frit is ground in a ball mill and mixed with concentrated sulphuric acid to form a slurry, which is sprayed in the form of a jet into a directly heated, rotating sulphating mill. The beryllium, now in a water-soluble form, is leached from the sludge, and ammonium hydroxide is added to the leach liquor, which is then fed to a crystallizer where ammonium alum is crystallized out. Chelating agents are added to the liquor to hold iron and nickel in solution, sodium hydroxide is then added, and the sodium beryllate thus formed is hydrolyzed to precipitate beryllium hydroxide. The latter product may be converted to beryllium fluoride for reduction by magnesium to metallic beryllium, or to beryllium chloride for electrolytic reduction.
In the fluoride process (figure 1) a briquetted mixture of ground ore, sodium silicofluoride and soda ash is sintered in a rotating hearth furnace. The sintered material is crushed, milled and leached. Sodium hydroxide is added to the solution of beryllium fluoride thus obtained and the precipitate of beryllium hydroxide is filtered in a rotary filter. Metallic beryllium is obtained as in the previous process by the magnesium reduction of beryllium fluoride or by electrolysis of beryllium chloride.
Figure 1. Production of beryllium oxide by the fluoride process
Uses
Beryllium is used in alloys with a number of metals including steel, nickel, magnesium, zinc and aluminium, the most widely used alloy being beryllium-copper—properly called “a bronze”—which has a high tensile strength and a capacity for being hardened by heat treatment. Beryllium bronzes are used in non-spark tools, electrical switch parts, watch springs, diaphragms, shims, cams and bushings.
One of the largest uses of the metal is as a moderator of thermal neutrons in nuclear reactors and as a reflector to reduce the leakage of neutrons from the reactor core. A mixed uranium-beryllium source is often used as a neutron source. As a foil, beryllium is used as window material in x-ray tubes. Its lightness, high elastic modulus and heat stability make it an attractive material for the aircraft and aerospace industry.
Beryllium oxide is made by heating beryllium nitrate or hydroxide.
It is used in the manufacture of ceramics, refractory materials and other beryllium compounds. It was used for the manufacture of phosphors for fluorescent lamps until the incidence of beryllium disease in the industry caused its use for this purpose to be abandoned (in 1949 in the United States).
Hazards
Fire and health hazards are associated with processes involving beryllium. Finely divided beryllium powder will burn, the degree of combustibility being a function of particle size. Fires have occurred in dust filtration units and during the welding of ventilation ducting in which finely divided beryllium was present.
Beryllium and its compounds are highly toxic substances. Beryllium can affect all organ systems, although the primary organ involved is the lung. Beryllium causes systemic disease by inhalation and can distribute itself widely throughout the body after absorption from the lungs. Little beryllium is absorbed from the gastro-intestinal tract. Beryllium can cause skin irritation and its traumatic introduction into subcutaneous tissue can cause local irritation and granuloma formation.
Pathogenesis
Beryllium in all its forms, except for beryl ore, has been associated with disease. The route of entry is by inhalation and in the acute disease there is a direct toxic effect on both the nasopharyngeal mucosa and that of the entire tracheobronchial tree as well, causing oedema and inflammation. In the lung it causes an acute chemical pneumonitis. The major form of beryllium toxicity at this point in time is chronic beryllium disease. A beryllium-specific delayed type of hypersensitivity is the major pathway of chronic disease. The entry of beryllium into the system through the lungs leads to proliferation of specific CD+ lymphocytes, with beryllium acting as a specific antigen, either alone or as a hapten through an interleukin-2 (IL2) receptor pathway. Individual susceptibility to beryllium thus can be explained on the basis of the individual CD+ response. Release of lymphokines from the activated lymphocytes then can lead to granuloma formation and macrophage recruitment. Beryllium can be transported to sites outside the lung where it can cause granuloma formation. Beryllium is released slowly from different sites and it is excreted by the kidneys. This slow release can occur over a span of 20 to 30 years. The chronicity and latency of disease can probably be explained on the basis of the slow metabolism and release phenomenon. The immune mechanisms involved in the pathogenesis of beryllium disease also allow for specific approaches to diagnosis, which will be discussed below.
Histopathology
The primary pathological finding in beryllium disease is the formation of non-caseating granulomas in the lungs, lymph nodes and at other sites. Histopathological studies of lungs in patients with acute beryllium disease have shown a non-specific pattern of acute and subacute bronchitis and pneumonitis. In chronic beryllium disease, there are varying degrees of lymphocytic infiltration of the lung interstitium and non-caseating granuloma formation (figure 2).
Figure 2. Lung tissue in a patient with chronic beryllium disease
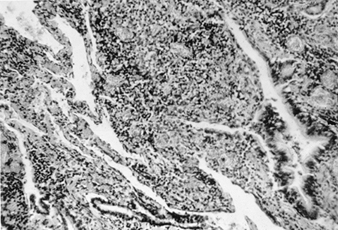
Both granulomas and round cell infiltration are visible
Many of the granulomas are located in the peribronchiolar areas. In addition, there can be histiocytes, plasma cells and giant cells with calcific inclusion bodies. If it is a case solely of granuloma formation, the long-term prognosis is better. The histology of the lung in chronic beryllium disease is indistinguishable from that of sarcoidosis. Non-caseating granulomas are also found in lymph nodes, liver, spleen, muscle and skin.
Clinical Manifestations
Skin injuries
Acid salts of beryllium cause allergic contact dermatitis. Such lesions may be erythematous, papular or papulovesicular, are commonly pruritic, and are found on exposed parts of the body. There is usually a delay of 2 weeks from first exposure to occurrence of the dermatitis, except in the case of heavy exposures, when an irritant reaction may be immediate. This delay is regarded as the time required to develop the hypersensitive state.
Accidental implantation of beryllium metal or crystals of a soluble beryllium compound in an abrasion, a crack in the skin or under the nail may cause an indurated area with central suppuration. Granulomas can also form at such sites.
Conjunctivitis and dermatitis may occur alone or together. In cases of conjunctivitis, periorbital oedema may be severe.
Acute disease
Beryllium nasopharyngitis is characterized by swollen and hyperaemic mucous membranes, bleeding points, fissures and ulceration. Perforation of the nasal septum has been described. Removal from exposure results in reversal of this inflammatory process within 3 to 6 weeks.
Involvement of the trachea and bronchial tree following exposure to higher levels of beryllium causes non-productive cough, substernal pain and moderate shortness of breath. Rhonchi and/or rales may be audible, and the x ray of the chest may show increased bronchovascular markings. The character and speed of onset and the severity of these signs and symptoms depend on the quality and quantity of exposure. Recovery is to be expected within 1 to 4 weeks if the worker is removed from further exposure.
The use of steroids is quite useful in countering the acute disease. No new cases of acute disease have been reported to the US Beryllium Case Registry in over 30 years. The Registry, which was started by Harriet Hardy in 1952, has almost 1,000 case records, among which are listed 212 acute cases. Almost all of these occurred in the fluorescent lamp manufacturing industry. Forty-four subjects with the acute disease subsequently developed chronic disease.
Chronic beryllium disease
Chronic beryllium disease is a pulmonary and systemic granulomatous disease caused by inhalation of beryllium. The latency of the disease can be from 1 to 30 years, most commonly occurring 10 to 15 years after first exposure. Chronic beryllium disease has a variable course with exacerbations and remissions in its clinical manifestations. However, the disease is usually progressive. There have been a few cases with chest x-ray abnormalities with a stable clinical course and without significant symptoms.
Exertional dyspnoea is the most common symptom of chronic beryllium disease. Other symptoms are cough, fatigue, weight loss, chest pain and arthralgias. Physical findings may be entirely normal or may include bibasilar crackles, lymphadenopathy, skin lesions, hepatosplenomegaly and clubbing. Signs of pulmonary hypertension may be present in severe, long-standing disease.
Renal stones and hyperuricaemia can occur in some patients and there have been rare reports of parotid gland enlargement and central nervous system involvement. The clinical manifestations of chronic beryllium disease are very similar to those of sarcoidosis.
Roentgenologic features
The x-ray pattern in chronic beryllium disease is non-specific and is similar to that which may be observed in sarcoidosis, idiopathic pulmonary fibrosis, tuberculosis, mycoses and dust disease (figure 3). Early in the course of the disease films may show granular, nodular or linear densities. These abnormalities may increase, decrease or remain unchanged, with or without fibrosis. Upper-lobe involvement is common. Hilar adenopathy, seen in approximately one-third of patients, is usually bilateral and accompanied by mottling of the lung fields. The absence of lung changes in the presence of adenopathy is a relative but not an absolute differential consideration in favour of sarcoidosis as opposed to chronic beryllium disease. Unilateral hilar adenopathy has been reported, but is quite rare.
Figure 3. Chest roentgenograph of a patient with chronic beryllium disease, showing diffuse fibronodular infiltrates and prominent hila
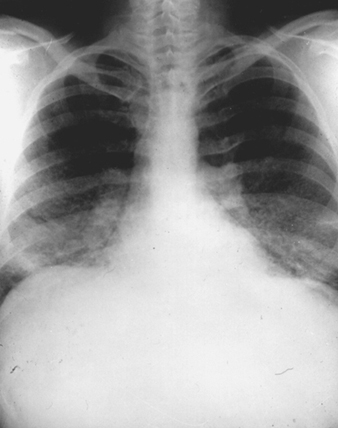
The x-ray picture does not correlate well with clinical status and does not reflect particular qualitative or quantitative aspects of the causal exposure.
Pulmonary function tests
Data from the Beryllium Case Registry show that 3 patterns of impairment may be found in chronic beryllium disease. Of 41 patients studied over a period of an average of 23 years after initial beryllium exposure, 20% had a restrictive defect, 36% had an interstitial defect (normal lung volumes and air flow rates but reduced diffusing capacity for carbon monoxide), 39% had an obstructive defect and 5% were normal. The obstructive pattern, which occurred in both smokers and non-smokers, was associated with granulomas in the peribronchial region. This study indicated that the pattern of impairment affects prognosis. Patients with interstitial defect fared best, with the least deterioration over a five-year interval. Patients with obstructive and restrictive defects experienced worsening of their impairment in spite of corticosteroid therapy.
Studies of lung function in beryllium extraction workers who were asymptomatic showed the presence of mild arterial hypoxaemia. This occurred usually within the first 10 years of exposure. In workers exposed to beryllium for 20 years or more there was a reduction in the forced vital capacity (FVC) and the forced expiratory volume in one second (FEV1). These findings suggest that the initial mild hypoxaemia could be due to the early alveolitis and that with further exposure and elapse of time the reduction in FEV1 and FVC could represent fibrosis and granuloma formation.
Other laboratory tests
Non-specific abnormal laboratory tests have been reported in chronic beryllium disease and include elevated sedimentation rate, erythrocytosis, increased gammaglobulin levels, hyperuricaemia and hypercalcaemia.
The Kveim skin test is negative in beryllium disease, whereas it may be positive in sarcoidosis. The angiotensin converting enzyme (ACE) level is usually normal in beryllium disease, but can be increased in 60% or more of patients with active sarcoidosis.
Diagnosis
Diagnosis of chronic beryllium disease for many years was based on the criteria developed through the Beryllium Case Registry, which included:
- a history of significant beryllium exposure
- evidence of lower respiratory tract disease
- abnormal chest x ray with interstitial fibronodular disease
- abnormal lung function tests with decreased carbon monoxide diffusing capacity (DLCO)
- pathological changes consistent with beryllium exposure in lung or thoracic lymph nodes
- the presence of beryllium in tissue.
Four of the six criteria had to be met and should have included either (1) or (6). Since the 1980s, advances in immunology have made it possible to make the diagnosis of beryllium disease without requiring tissue specimens for histological examination or beryllium analysis. The transformation of lymphocytes in blood in response to beryllium exposure (as in the lymphocyte transformation test, LTT) or lymphocytes from bronchoalveolar lavage (BAL) have been proposed by Newman et al. (1989) as useful diagnostic tools in making the diagnosis of beryllium disease in exposed subjects. Their data suggest that a positive blood LTT is indicative of sensitization. However, recent data show that the blood LTT does not correlate well with pulmonary disease. The BAL lymphocyte transformation correlates much better with abnormal pulmonary function and does not correlate well with concurrent abnormalities in the blood LTT. Thus, to make a diagnosis of beryllium disease, one needs a combination of clinical, radiological and lung function abnormalities and a positive LTT in the BAL. A positive blood LTT by itself is not diagnostic. Microprobe analysis of small tissue samples for beryllium is another recent innovation which could help in diagnosis of disease in small lung tissue samples obtained by transbronchial lung biopsy.
Sarcoidosis is the disorder most closely resembling chronic beryllium disease, and the differentiation may be difficult. Thus far, no cystic bone disease or involvement of the eye or tonsil has appeared in chronic beryllium disease. Similarly, the Kveim test is negative in beryllium disease. Skin testing to demonstrate beryllium sensitization is not recommended, in that the test itself is sensitizing, may possibly trigger systemic reactions in sensitized people and does not of itself establish that the presenting disease is necessarily beryllium related.
More sophisticated immunological approaches in differential diagnosis should allow for better differentiation from sarcoidosis in the future.
Prognosis
The prognosis of chronic beryllium disease has altered favourably during the years; it has been suggested that the longer delays in onset observed among beryllium workers may reflect lower exposure or lower beryllium body burden, resulting in a milder clinical course. Clinical evidence is that steroid therapy, if used when measurable disability first appears, in adequate doses for long enough periods, has improved the clinical status of many patients, allowing some of them to return to useful jobs. There is no clear evidence that steroids have cured chronic beryllium poisoning.
Beryllium and cancer
In animals, experimentally administered beryllium is a carcinogen, causing osteogenic sarcoma after intravenous injection in rabbits and lung cancer after inhalation in rats and monkeys. Whether beryllium may be a human carcinogen is a controversial issue. Some epidemiological studies have suggested an association, particularly after acute beryllium disease. This finding has been disputed by others. One can conclude that beryllium is carcinogenic in animals and there may be a link between lung cancer and beryllium in humans, particularly in those with the acute disease.
Safety and Health Measures
Safety and health precautions must cover the fire hazard as well as the much more serious toxicity danger.
Fire prevention
Arrangements must be made to prevent possible sources of ignition, such as the sparking or arcing of electrical apparatus, friction, and so forth, in the vicinity of finely divided beryllium powder. Equipment in which this powder has been present should be emptied and cleaned before acetylene or electrical welding apparatus is used on it. Oxide-free, ultrafine beryllium powder that has been prepared in inert gas is liable to ignite spontaneously on exposure to air.
Suitable dry powder—not water—should be used to extinguish a beryllium fire. Full personal protective equipment, including respiratory protective equipment, should be worn and firefighters should bathe afterwards and arrange for their clothing to be laundered separately.
Health protection
Beryllium processes must be conducted in a carefully controlled manner to protect both the worker and the general population. The main risk takes the form of airborne contamination and the process and plant should be designed to give rise to as little dust or fume as possible. Wet processes should be used instead of dry processes, and the ingredients of beryllium-containing preparations should be unified as aqueous suspensions instead of as dry powders; whenever possible the plant should be designed as groups of separate enclosed units. The permissible concentration of beryllium in the atmosphere is so low that enclosure must be applied even to wet processes, otherwise escaping splashes and spills can dry out and the dust can enter the atmosphere.
Operations from which dust may be evolved should be conducted in areas with maximum degree of enclosure consistent with the needs of manipulation. Some operations are performed in glove boxes, but many more are conducted in enclosures provided with exhaust ventilation similar to that installed in chemical fume cupboards. Machining operations may be ventilated by high-velocity, low-volume local exhaust systems or by hooded enclosures with exhaust ventilation.
To check the effectiveness of these precautionary measures atmosphere monitoring should be done in such a manner that the daily average exposure of workers to respirable beryllium can be calculated. The work area should be cleaned regularly by means of a proper vacuum cleaner or a wet mop. Beryllium processes should be segregated from the other operations in the factory.
Personal protective equipment should be provided for workers engaged in beryllium processes. Where they are fully employed in processes involving the manipulation of beryllium compounds or in processes associated with the extraction of the metal from the ore, provision should be made for a complete change of clothing so that the workers do not go home wearing clothing in which they have been working. Arrangements should be made for the safe laundering of such working clothes, and protective overalls should be provided even to laundry workers to ensure that they too are not exposed to risk. These arrangements should not be left to normal home laundering procedures. Cases of beryllium poisoning in the families of workers have been attributed to workers taking contaminated clothing home or wearing them in the home.
An occupational health standard of 2μg/m3, proposed in 1949 by a committee operating under the auspices of the US Atomic Energy Commission, continues to be widely observed. Existing interpretations generally permit fluctuations to a “ceiling” of 5μg/m3 as long as the time-weighted average is not exceeded. Additionally, an “acceptable maximum peak above the ceiling concentration for an eight-hour shift” of 25μg/m3 for up to 30 min is also permissible. These operational levels are achievable in current industrial practice, and there is no evidence of adverse health experience among persons working in an environment thus controlled. Because of a possible link between beryllium and lung cancer it has been suggested that the allowable limit be reduced to 1μg/m3, but no official action has been taken on this suggestion in the United States.
The population at risk for developing beryllium disease is that which in some manner deals with beryllium in its extraction or subsequent use. However, a few “neighbourhood” cases have been reported from a distance 1 to 2 km from beryllium extraction plants.
Pre-employment and periodical medical examinations of workers exposed to beryllium and its compounds are compulsory in a number of countries. Recommended evaluation includes an annual respiratory questionnaire, a chest x ray and lung function tests. With advances in immunology, the LTT may also become a routine evaluation, although at this time not enough data are available to recommend its use routinely. With evidence of beryllium disease, it is unwise to allow a worker to be exposed to beryllium further, even though the workplace meets the threshold criteria for beryllium concentration in the air.
Treatment
The major step in therapy is avoidance of further exposure to beryllium. Corticosteroids are the primary mode of therapy in chronic beryllium disease. Corticosteroids appear to alter the course of disease favourably but do not “cure” it.
Corticosteroids should be started on a daily basis with a relatively high dose of Prednisone of 0.5 to 1 mg per kg or more, and continued until improvement occurs or no further deterioration in clinical or lung function tests occurs. Usually this takes 4 to 6 weeks. Slow reduction of steroids is recommended, and eventually alternate-day therapy may be possible. Steroid therapy ordinarily becomes a lifelong necessity.
Other supportive measures such as supplemental oxygen, diuretics, digitalis and antibiotics (when infection exists) are indicated as the clinical condition of the patient would dictate. Immunization against influenza and pneumococcus should also be considered, as with any patient with chronic respiratory disease.
Diseases Caused by Organic Dusts
Organic Dust and Disease
Dusts of vegetable, animal and microbial origin have always been part of the human environment. When the first aquatic organisms moved to land some 450 million years ago, they soon developed defence systems against the many noxious substances present in the terrestrial environment, most of them of plant origin. Exposures to this environment usually cause no specific problems, even though plants contain a number of extremely toxic substances, particularly those present in or produced by moulds.
During the development of civilization, climatic conditions in some parts of the world necessitated certain activities to be undertaken indoors. Threshing in the Scandinavian countries was performed indoors during the winter, a practice mentioned by chroniclers in antiquity. The enclosure of dusty processes led to disease among the exposed persons, and one of the first published accounts of this is by the Danish bishop Olaus Magnus (1555, as cited by Rask-Andersen 1988). He described a disease among threshers in Scandinavia as follows:
“In separating the grain from the chaff, care must be taken to choose a time when there is a suitable wind which will sweep away the grain dust, so that it will not damage the vital organs of the threshers. This dust is so fine that it will almost unnoticeably penetrate into the mouth and accumulate in the throat. If this is not quickly dealt with by drinking fresh ale, the thresher may never again or only for a short period eat what he has threshed.”
With the introduction of machine processing of organic materials, treatment of large quantities of materials indoors with poor ventilation led to high levels of airborne dust. The descriptions by bishop Olaus Magnus and later by Ramazzini (1713) were followed by several reports on disease and organic dusts in the nineteenth century, particularly among cotton mill workers (Leach 1863; Prausnitz 1936). Later, the specific pulmonary disease common among farmers handling mouldy materials was also described (Campbell 1932).
During recent decades, a large number of reports on disease among persons exposed to organic dusts have been published. Initially, most of these were based on persons seeking medical help. The names of the diseases, when published, were often related to the particular environment where the disease was first recognized, and a bewildering array of names resulted, such as farmer’s lung, mushroom grower’s lung, brown lung and humidifier fever.
With the advent of modern epidemiology, more reliable figures were obtained for the incidence of occupational respiratory diseases related to organic dust (Rylander, Donham and Peterson 1986; Rylander and Peterson 1990). There was also advancement in the understanding of the pathological mechanisms underlying these diseases, particularly the inflammatory response (Henson and Murphy 1989). This paved the way for a more coherent picture of diseases caused by organic dusts (Rylander and Jacobs 1997).
The following will describe the different organic dust environments where disease has been reported, the disease entities themselves, the classical byssinosis disease and specific preventive measures.
Environments
Organic dusts are airborne particles of vegetable, animal or microbial origin. Table 1 lists examples of environments, work processes and agents involving the risk of exposure to organic dusts.
Table 1. Examples of sources of hazards of exposure to organic dust
Agriculture
Handling of grain, hay or other crops
Sugar-cane processing
Greenhouses
Silos
Animals
Swine/dairy confinement buildings
Poultry houses and processing plants
Laboratory animals, farm animals and pets
Waste-processing
Sewage water and silt
Household garbage
Composting
Industry
Vegetable fibre processing (cotton, flax, hemp, jute, sisal)
Fermentation
Timber and wood processing
Bakeries
Biotechnology processing
Buildings
Contaminated water in humidifiers
Microbial growth on structures or in ventilation ducts
Agents
It is now understood that the specific agents in the dusts are the major reason why disease develops. Organic dusts contain a multitude of agents with potential biological effects. Some of the major agents are found in table 2.
Table 2. Major agents in organic dusts with potential biological activity
Vegetable agents
Tannins
Histamine
Plicatic acid
Alkaloids (e.g., nicotine)
Cytochalasins
Animal agents
Proteins
Enzymes
Microbial agents
Endotoxins
(1→3)–β–D-glucans
Proteases
Mycotoxins
The relative role of each of these agents, alone or in combination with others, for the development of disease, is mostly unknown. Most of the information available relates to bacterial endotoxins which are present in all organic dusts.
Endotoxins are lipopolysaccharide compounds which are attached to the outer cell surface of Gram-negative bacteria. Endotoxin has a wide variety of biological properties. After inhalation it causes an acute inflammation (Snella and Rylander 1982; Brigham and Meyrick 1986). An influx of neutrophils (leukocytes) into the lung and the airways is the hallmark of this reaction. It is accompanied by activation of other cells and secretion of inflammatory mediators. After repeated exposures, the inflammation decreases (adaptation). The reaction is limited to the airway mucosa, and there is no extensive involvement of the lung parenchyma.
Another specific agent in organic dust is (1→3)-β-D-glucan. This is a polyglucose compound present in the cell wall structure of moulds and some bacteria. It enhances the inflammatory response caused by endotoxin and alters the function of inflammatory cells, particularly macrophages and T-cells (Di Luzio 1985; Fogelmark et al. 1992).
Other specific agents present in organic dusts are proteins, tannins, proteases and other enzymes, and toxins from moulds. Very little data are available on the concentrations of these agents in organic dusts. Several of the specific agents in organic dusts, such as proteins and enzymes, are allergens.
Diseases
The diseases caused by organic dusts are shown in table 3 with the corresponding International Classification of Disease (ICD) numbers (Rylander and Jacobs 1994).
Table 3. Diseases induced by organic dusts and their ICD codes
Bronchitis and pneumonitis (ICD J40)
Toxic pneumonitis (inhalation fever, organic dust toxic syndrome)
Airways inflammation (mucous membrane inflammation)
Chronic bronchitis (ICD J42)
Hypersensitivity pneumonitis (allergic alveolitis) (ICD J67)
Asthma (ICD J45)
Rhinitis, conjunctivitis
The primary route of exposure for organic dusts is by inhalation, and consequently the effects on the lung have received the major share of attention in research as well as in clinical work. There is, however, a growing body of evidence from published epidemiological studies and case reports as well as anecdotal reports, that systemic effects also occur. The mechanism involved seems to be a local inflammation at the target site, the lung, and a subsequent release of cytokines either with systemic effects (Dunn 1992; Michel et al. 1991) or an effect on the epithelium in the gut (Axmacher et al. 1991). Non-respiratory clinical effects are fever, joint pains, neurosensory effects, skin problems, intestinal disease, fatigue and headache.
The different disease entities as described in table 3 are easy to diagnose in typical cases, and the underlying pathology is distinctly different. In real life, however, a worker who has a disease due to organic dust exposure, often presents a mixture of the different disease entities. One person may have airways inflammation for a number of years, suddenly develop asthma and in addition have symptoms of toxic pneumonitis during a particularly heavy exposure. Another person may have subclinical hypersensitivity pneumonitis with lymphocytosis in the airways and develop toxic pneumonitis during a particularly heavy exposure.
A good example of the mixture of disease entities that may appear is byssinosis. This disease was first described in the cotton mills, but the individual disease entities are also found in other organic dust environments. An overview of the disease follows.
Byssinosis
The disease
Byssinosis was first described in the 1800s, and a classic report involving clinical as well as experimental work was given by Prausnitz (1936). He described the symptoms among cotton mill workers as follows:
“After working for years without any appreciable trouble except a little cough, cotton mill workers notice either a sudden aggravation of their cough, which becomes dry and exceedingly irritating¼ These attacks usually occur on Mondays ¼ but gradually the symptoms begin to spread over the ensuing days of the week; in time the difference disappears and they suffer continuously.”
The first epidemiological investigations were performed in England in the 1950s (Schilling et al. 1955; Schilling 1956). The initial diagnosis was based on the appearance of a typical Monday morning chest tightness, diagnosed using a questionnaire (Roach and Schilling 1960). A scheme for grading the severity of byssinosis based on the type and periodicity of symptoms was developed (Mekky, Roach and Schilling 1967; Schilling et al. 1955). Duration of exposure was used as a measure of dose and this was related to the severity of the response. Based on clinical interviews of large numbers of workers, this grading scheme was later modified to more accurately reflect the time intervals for the decrease in FEV1 (Berry et al. 1973).
In one study, a difference in the prevalence of byssinosis in mills processing different types of cotton was found (Jones et al. 1979). Mills using high-quality cotton to produce finer yarns had a lower prevalence of byssinosis than mills producing coarse yarns and using a lower quality of cotton. Thus in addition to exposure intensity and duration, both dose-related variables, the type of dust became an important variable for assessing exposure. Later it was demonstrated that the differences in the response of workers exposed to coarse and medium cottons was dependent not only on the type of cotton but on other variables that affect exposure, including: processing variables such as carding speed, environmental variables such as humidification and ventilation, and manufacturing variables such as different yarn treatments (Berry et al. 1973).
The next refinement of the relationship between exposure to cotton dust and a response (either symptoms or objective measures of pulmonary function), was the studies from the United States, comparing those who worked in 100% cotton to workers using the same cotton but in a 50:50 blend with synthetics and workers without exposure to cotton (Merchant et al. 1973). Workers exposed to 100% cotton had the highest prevalence of byssinosis independent of cigarette smoking, one of the confounders of exposure to cotton dust. This semiquantitative relationship between dose and response to cotton dust was further refined in a group of textile workers stratified by sex, smoking, work area and mill type. A relationship was observed in each of these categories between dust concentration in the lower dust ranges and byssinosis prevalence and/or change in forced expiratory volume in one second (FEV1).
In later investigations, the FEV1 decrease over the work shift has been used to assess the effects of exposure, and it is also a part of the US Cotton Dust Standard.
Byssinosis was long regarded as a peculiar disease with a mixture of different symptoms and no knowledge of the specific pathology. Some authors suggested that it was an occupational asthma (Bouhuys 1976). A workgroup meeting in 1987 analysed the symptomatology and pathology of the disease (Rylander et al. 1987). It was agreed that the disease comprised several clinical entities, generally related to organic dust exposure.
Toxic pneumonitis may appear the first time an employee works in the mill, particularly when working in the opening, blowing and carding sections (Trice 1940). Although habituation develops, the symptoms may reappear after an unusually heavy exposure later on.
Airways inflammation is the most widespread disease, and it appears at different degrees of severity from light irritation in the nose and airways to severe dry cough and breathing difficulties. The inflammation causes constriction of airways and a reduced FEV1. Airway responsiveness is increased as measured with a methacholine or histamine challenge test. It has been discussed whether airways inflammation should be accepted as a disease entity by itself or whether it merely represents a symptom. As the clinical findings in terms of severe cough with airways narrowing can lead to a decrease in work ability, it is justified to regard it as an occupational disease.
Continued airways inflammation over several years may develop into chronic bronchitis, particularly among heavily exposed workers in the blowing and carding areas. The clinical picture would be one of chronic obstructive pulmonary disease (COPD).
Occupational asthma develops in a small percentage of the workforce, but is usually not diagnosed in cross-sectional studies as the workers are forced to leave work because of the disease. Hypersensitivity pneumonitis has not been detected in any of the epidemiological studies undertaken, nor have there been case reports relating to cotton dust exposure. The absence of hypersensitivity pneumonitis may be due to the relatively low amount of moulds in cotton, as mouldy cotton is not acceptable for processing.
A subjective feeling of chest tightness, most common on Mondays, is the classical symptom of cotton dust exposure (Schilling et al. 1955). It is not, however, a feature unique to cotton dust exposure as it appears also among persons working with other kinds of organic dusts (Donham et al. 1989). Chest tightness develops slowly over a number of years but it can also be induced in previously unexposed persons, provided that the dose level is high (Haglind and Rylander 1984). The presence of chest tightness is not directly related to a decrease in FEV1.
The pathology behind chest tightness has not been explained. It has been suggested that the symptoms are due to an increased adhesiveness of platelets which accumulate in the lung capillaries and increase the pulmonary artery pressure. It is likely that chest tightness involves some kind of cell sensitization, as it takes repeated exposures for the symptom to develop. This hypothesis is supported by results from studies on blood monocytes from cotton workers (Beijer et al. 1990). A higher ability to produce procoagulant factor, indicative of cell sensitization, was found among cotton workers as compared to controls.
The environment
The disease was originally described among workers in cotton, flax and soft hemp mills. In the first phase of cotton treatment within the mills—bale opening, blowing and carding—more than half of the workers may have symptoms of chest tightness and airways inflammation. The incidence decreases as the cotton is processed, reflecting the successive cleaning of the causative agent from the fibre. Byssinosis has been described in all countries where investigations in cotton mills have been performed. Some countries like Australia have, however, unusually low incidence figures (Gun et al. 1983).
There is now uniform evidence that bacterial endotoxins are the causative agent for toxic pneumonitis and airways inflammation (Castellan et al. 1987; Pernis et al. 1961; Rylander, Haglind and Lundholm 1985; Rylander and Haglind 1986; Herbert et al. 1992; Sigsgaard et al. 1992). Dose-response relationships have been described and the typical symptoms have been induced by inhalation of purified endotoxin (Rylander et al. 1989; Michel et al. 1995). Although this does not exclude the possibility that other agents could contribute to the pathogenesis, endotoxins can serve as markers for disease risk. It is unlikely that endotoxins are related to the development of occupational asthma, but they could act as an adjuvant for potential allergens in cotton dust.
The case
The diagnosis of byssinosis is classically made using questionnaires with the specific question “Does your chest feel tight, and if so, on which day of the week?”. Persons with Monday morning chest tightness are classified as byssinotics according to a scheme suggested by Schilling (1956). Spirometry can be performed, and, according to the different combinations of chest tightness and decrease in FEV1, the diagnostic scheme illustrated in table 4 has evolved.
Table 4. Diagnostic criteria for byssinosis
Grade ½. Chest tightness on the first day of some working weeks
Grade 1. Chest tightness on the first day of every working week
Grade 2. Chest tightness on the first and other days of the working week
Grade 3. Grade 2 symptoms accompanied by evidence of permanent incapacity in the form of diminished effort intolerance and/or reduced ventilatory capacity
Treatment
Treatment in the light stages of byssinosis is symptomatic, and most of the workers learn to live with the slight chest tightness and bronchoconstriction that they experience on Mondays or when cleaning machinery or carrying out similar tasks with a higher than normal exposure. More advanced stages of airways inflammation or regular chest tightness several days of the week require transfer to less dusty operations. The presence of occupational asthma mostly requires work change.
Prevention
Prevention in general is dealt with in detail elsewhere in the Encyclopaedia. The basic principles for prevention in terms of product substitute, exposure limitation, worker protection and screening for disease apply also for cotton dust exposure.
Regarding product substitutes, it has been suggested that cotton with a low level of bacterial contamination be used. An inverse proof of this concept is found in reports from 1863 where the change to dirty cotton provoked an increase in the prevalence of symptoms among the exposed workers (Leach 1863). There is also the possibility of changing to other fibres, particularly synthetic fibres, although this is not always feasible from a product point of view. There is at present no production-applied technique to decrease the endotoxin content of cotton fibres.
Regarding dust reduction, successful programmes have been implemented in the United States and elsewhere (Jacobs 1987). Such programmes are expensive, and the costs for highly efficient dust removal may be prohibitive for developing countries (Corn 1987).
Regarding exposure control, the level of dust is not a sufficiently precise measure of exposure risk. Depending on the degree of contamination with Gram-negative bacteria and thus endotoxin, a given dust level may or may not be associated with a risk. For endotoxins, no official guidelines have been established. It has been suggested that a level of 200 ng/m3 is the threshold for toxic pneumonitis, 100 to 200 ng/m3 for acute airways constriction over the workshift and 10 ng/m3 for airways inflammation (Rylander and Jacobs 1997).
Knowledge about the risk factors and the consequences of exposure are important for prevention. The information basis has expanded rapidly during recent years, but much of it is not yet present in textbooks or other easily available sources. A further problem is that symptoms and findings in respiratory diseases induced by organic dust are non-specific and occur normally in the population. They may thus not be correctly diagnosed in the early stages.
Proper dissemination of knowledge concerning the effects of cotton and other organic dusts requires the establishment of appropriate training programmes. These should be directed not only towards workers with potential exposure but also towards employers and health personnel, particularly occupational health inspectors and engineers. Information must include source identification, symptoms and disease description, and methods of protection. An informed worker can more readily recognize work-related symptoms and communicate more effectively to a health care provider. Regarding health surveillance and screening, questionnaires are a major instrument to be used. Several versions of questionnaires specifically designed for diagnosing diseases induced by organic dust have been reported in the literature (Rylander, Peterson and Donham 1990; Schwartz et al. 1995). Lung function testing is also a useful tool for surveillance and diagnosis. Measurements of airway responsiveness have been found to be useful (Rylander and Bergström 1993; Carvalheiro et al. 1995). Other diagnostic tools such as measurements of inflammatory mediators or cell activity are still in the research phase.
Occupational Asthma
Asthma is a respiratory disease characterized by airway obstruction that is partially or completely reversible, either spontaneously or with treatment; airway inflammation; and increased airway responsiveness to a variety of stimuli (NAEP 1991). Occupational asthma (OA) is asthma that is caused by environmental exposures in the workplace. Several hundred agents have been reported to cause OA. Pre-existing asthma or airway hyper-responsiveness, with symptoms worsened by work exposure to irritants or physical stimuli, is usually classified separately as work-aggravated asthma (WAA). There is general agreement that OA has become the most prevalent occupational lung disease in developed countries, although estimates of actual prevalence and incidence are quite variable. It is clear, however, that in many countries asthma of occupational aetiology causes a largely unrecognized burden of disease and disability with high economic and non-economic costs. Much of this public health and economic burden is potentially preventable by identifying and controlling or eliminating the workplace exposures causing the asthma. This article will summarize current approaches to recognition, management and prevention of OA. Several recent publications discuss these issues in more detail (Chan-Yeung 1995; Bernstein et al. 1993).
Magnitude of the Problem
Prevalences of asthma in adults generally range from 3 to 5%, depending on the definition of asthma and geographic variations, and may be considerably higher in some low-income urban populations. The proportion of adult asthma cases in the general population that is related to the work environment is reported to range from 2 to 23%, with recent estimates tending towards the higher end of the range. Prevalences of asthma and OA have been estimated in small cohort and cross-sectional studies of high-risk occupational groups. In a review of 22 selected studies of workplaces with exposures to specific substances, prevalences of asthma or OA, defined in various ways, ranged from 3 to 54%, with 12 studies reporting prevalences over 15% (Becklake, in Bernstein et al. 1993). The wide range reflects real variation in actual prevalence (due to different types and levels of exposure). It also reflects differences in diagnostic criteria, and variation in the strength of the biases, such as “survivor bias” which may result from exclusion of workers who developed OA and left the workplace before the study was conducted. Population estimates of incidence range from 14 per million employed adults per year in the United States to 140 per million employed adults per year in Finland (Meredith and Nordman 1996). Ascertainment of cases was more complete and methods of diagnosis were generally more rigorous in Finland. The evidence from these different sources is consistent in its implication that OA is often under-diagnosed and/or under-reported and is a public health problem of greater magnitude than generally recognized.
Causes of Occupational Asthma
Over 200 agents (specific substances, occupations or industrial processes) have been reported to cause OA, based on epidemiological and/or clinical evidence. In OA, airway inflammation and bronchoconstriction can be caused by immunological response to sensitizing agents, by direct irritant effects, or by other non-immunological mechanisms. Some agents (e.g., organophosphate insecticides) may also cause bronchoconstriction by direct pharmacological action. Most of the reported agents are thought to induce a sensitization response. Respiratory irritants often worsen symptoms in workers with pre-existing asthma (i.e., WAA) and, at high exposure levels, can cause new onset of asthma (termed reactive airways dysfunction syndrome (RADS) or irritant-induced asthma) (Brooks, Weiss and Bernstein 1985; Alberts and Do Pico 1996).
OA may occur with or without a latency period. Latency period refers to the time between initial exposure and development of symptoms, and is highly variable. It is often less than 2 years, but in around 20% of cases is 10 years or longer. OA with latency is generally caused by sensitization to one or more agents. RADS is an example of OA without latency.
High molecular weight sensitizing agents (5,000 daltons (Da) or greater) often act by an IgE-dependent mechanism. Low molecular weight sensitizing agents (less than 5,000 Da), which include highly reactive chemicals like isocyanates, may act by IgE-independent mechanisms or may act as haptens, combining with body proteins. Once a worker becomes sensitized to an agent, re-exposure (frequently at levels far below the level that caused sensitization) results in an inflammatory response in the airways, often accompanied by increases in airflow limitation and non-specific bronchial responsiveness (NBR).
In epidemiological studies of OA, workplace exposures are consistently the strongest determinants of asthma prevalence, and the risk of developing OA with latency tends to increase with estimated intensity of exposure. Atopy is an important and smoking a somewhat less consistent determinant of asthma occurrence in studies of agents that act through an IgE-dependent mechanism. Neither atopy nor smoking appears to be an important determinant of asthma in studies of agents acting through IgE-independent mechanisms.
Clinical Presentation
The symptom spectrum of OA is similar to non-occupational asthma: wheeze, cough, chest tightness and shortness of breath. Patients sometimes present cough-variant or nocturnal asthma. OA can be severe and disabling, and deaths have been reported. Onset of OA occurs due to a specific job environment, so identifying exposures that occurred at the time of onset of asthmatic symptoms is key to an accurate diagnosis. In WAA, workplace exposures cause a significant increase in frequency and/or severity of symptoms of pre-existing asthma.
Several features of the clinical history may suggest occupational aetiology (Chan-Yeung 1995). Symptoms frequently worsen at work or at night after work, improve on days off, and recur on return to work. Symptoms may worsen progressively towards the end of the workweek. The patient may note specific activities or agents in the workplace that reproducibly trigger symptoms. Work-related eye irritation or rhinitis may be associated with asthmatic symptoms. These typical symptom patterns may be present only in the initial stages of OA. Partial or complete resolution on weekends or vacations is common early in the course of OA, but with repeated exposures, the time required for recovery may increase to one or two weeks, or recovery may cease to occur. The majority of patients with OA whose exposures are terminated continue to have symptomatic asthma even years after cessation of exposure, with permanent impairment and disability. Continuing exposure is associated with further worsening of asthma. Brief duration and mild severity of symptoms at the time of cessation of exposure are good prognostic factors and decrease the likelihood of permanent asthma.
Several characteristic temporal patterns of symptoms have been reported for OA. Early asthmatic reactions typically occur shortly (less than one hour) after beginning work or the specific work exposure causing the asthma. Late asthmatic reactions begin 4 to 6 hours after exposure begins, and can last 24 to 48 hours. Combinations of these patterns occur as dual asthmatic reactions with spontaneous resolution of symptoms separating an early and late reaction, or as continuous asthmatic reactions with no resolution of symptoms between phases. With exceptions, early reactions tend to be IgE mediated, and late reactions tend to be IgE independent.
Increased NBR, generally measured by methacholine or histamine challenge, is considered a cardinal feature of occupational asthma. The time course and degree of NBR may be useful in diagnosis and monitoring. NBR may decrease within several weeks after cessation of exposure, although abnormal NBR commonly persists for months or years after exposures are terminated. In individuals with irritant-induced occupational asthma, NBR is not expected to vary with exposure and/or symptoms.
Recognition and Diagnosis
Accurate diagnosis of OA is important, given the substantial negative consequences of either under- or over-diagnosis. In workers with OA or at risk of developing OA, timely recognition, identification and control of the occupational exposures causing the asthma improve the chances of prevention or complete recovery. This primary prevention can greatly reduce the high financial and human costs of chronic, disabling asthma. Conversely, since a diagnosis of OA may obligate a complete change of occupation, or costly interventions in the workplace, accurately distinguishing OA from asthma that is not occupational can prevent unnecessary social and financial costs to both employers and workers.
Several case definitions of OA have been proposed, appropriate in different circumstances. Definitions found valuable for worker screening or surveillance (Hoffman et al. 1990) may not be entirely applicable for clinical purposes or compensation. A consensus of researchers has defined OA as “a disease characterized by variable airflow limitation and/or airway hyper-responsiveness due to causes and conditions attributable to a particular occupational environment and not to stimuli encountered outside the workplace” (Bernstein et al. 1993). This definition has been operationalized as a medical case definition, summarized in table 1 (Chan-Yeung 1995).
Table 1. ACCP medical case definition of occupational asthma
Criteria for diagnosis of occupational asthma1 (requires all 4, A-D):
(A) Physician diagnosis of asthma and/or physiological evidence of airways hyper-responsiveness
(B) Occupational exposure preceded onset of asthmatic symptoms1
(C) Association between symptoms of asthma and work
(D) Exposure and/or physiological evidence of relation of asthma to workplace environment (Diagnosis of OA requires one or more of D2-D5, likely OA requires only D1)
(1) Workplace exposure to agent reported to give rise to OA
(2) Work-related changes in FEV1 and/or PEF
(3) Work-related changes in serial testing for non-specific bronchial responsiveness (e.g., Methacholine Challenge Test)
(4) Positive specific bronchial challenge test
(5) Onset of asthma with a clear association with a symptomatic exposure to an inhaled irritant in the workplace (generally RADS)
Criteria for diagnosis of RADS (should meet all 7):
(1) Documented absence of preexisting asthma-like complaints
(2) Onset of symptoms after a single exposure incident or accident
(3) Exposure to a gas, smoke, fume, vapour or dust with irritant properties present in high concentration
(4) Onset of symptoms within 24 hours after exposure with persistence of symptoms for at least 3 months
(5) Symptoms consistent with asthma: cough, wheeze, dyspnoea
(6) Presence of airflow obstruction on pulmonary function tests and/or presence of non-specific bronchial hyper-responsiveness (testing should be done shortly after exposure)
(7) Other pulmonary diseases ruled out
Criteria for diagnosis of work-aggravated asthma (WAA):
(1) Meets criteria A and C of ACCP Medical Case Definition of OA
(2) Pre-existing asthma or history of asthmatic symptoms, (with active symptoms during the year prior to start of employment or exposure of interest)
(3) Clear increase in symptoms or medication requirement, or documentation of work-related changes in PEFR or FEV1 after start of employment or exposure of interest
1 A case definition requiring A, C and any one of D1 to D5 may be useful in surveillance for OA, WAA and RADS.
Source: Chan-Yeung 1995.
Thorough clinical evaluation of OA can be time consuming, costly and difficult. It may require diagnostic trials of removal from and return to work, and often requires the patient to reliably chart serial peak expiratory flow (PEF) measurements. Some components of the clinical evaluation (e.g., specific bronchial challenge or serial quantitative testing for NBR) may not be readily available to many physicians. Other components may simply not be achievable (e.g., patient no longer working, diagnostic resources not available, inadequate serial PEF measurements). Diagnostic accuracy is likely to increase with the thoroughness of the clinical evaluation. In each individual patient, decisions on the extent of medical evaluation will need to balance costs of the evaluation with the clinical, social, financial and public health consequences of incorrectly diagnosing or ruling out OA.
In consideration of these difficulties, a stepped approach to diagnosis of OA is outlined in table 2. This is intended as a general guide to facilitate accurate, practical and efficient diagnostic evaluation, recognizing that some of the suggested procedures may not be available in some settings. Diagnosis of OA involves establishing both the diagnosis of asthma and the relation between asthma and workplace exposures. After each step, for each patient, the physician will need to determine whether the level of diagnostic certainty achieved is adequate to support the necessary decisions, or whether evaluation should continue to the next step. If facilities and resources are available, the time and cost of continuing the clinical evaluation are usually justified by the importance of making an accurate determination of the relationship of asthma to work. Highlights of diagnostic procedures for OA will be summarized; details can be found in several of the references (Chan-Yeung 1995; Bernstein et al. 1993). Consultation with a physician experienced in OA may be considered, since the diagnostic process may be difficult.
Table 2. Steps in diagnostic evaluation of asthma in the workplace
Step 1 Thorough medical and occupational history and directed physical examination.
Step 2 Physiologic evaluation for reversible airway obstruction and/or non specific bronchial hyper-responsiveness.
Step 3 Immunologic assessment, if appropriate.
Assess Work Status:
Currently working: Proceed to Step 4 first.
Not currently working, diagnostic trial of return to work feasible: Step 5 first, then Step 4.
Not currently working, diagnostic trial of return to work not feasible: Step 6.
Step 4 Clinical evaluation of asthma at work or diagnostic trial of return to work.
Step 5 Clinical evaluation of asthma away from work or diagnostic trial of removal from work.
Step 6 Workplace challenge or specific bronchial challenge testing. If available for suspected causal exposures, this step may be performed prior to Step 4 for any patient.
This is intended as a general guide to facilitate practical and efficient diagnostic evaluation. It is recommended that physicians who diagnose and manage OA refer to current clinical literature as well.
RADS, when caused by an occupational exposure, is usually considered a subclass of OA. It is diagnosed clinically, using the criteria in Table 6. Patients who have experienced significant respiratory injury due to high-level irritant inhalations should be evaluated for persistent symptoms and presence of airflow obstruction shortly after the event. If the clinical history is compatible with RADS, further evaluation should include quantitative testing for NBR, if not contra-indicated.
WAA may be common, and may cause a substantial preventable burden of disability, but little has been published on diagnosis, management or prognosis. As summarized in Table 6, WAA is recognized when asthmatic symptoms preceded the suspected causal exposure but are clearly aggravated by the work environment. Worsening at work can be documented either by physiological evidence or through evaluation of medical records and medication use. It is a clinical judgement whether patients with a history of asthma in remission, who have recurrence of asthmatic symptoms that otherwise meet the criteria for OA, are diagnosed with OA or WAA. One year has been proposed as a sufficiently long asymptomatic period that the onset of symptoms is likely to represent a new process caused by the workplace exposure, although no consensus yet exists.
Step 1: Thorough medical and occupational history anddirected physical examination
Initial suspicion of possible OA in appropriate clinical and workplace situations is key, given the importance of early diagnosis and intervention in improving prognosis. The diagnosis of OA or WAA should be considered in all asthmatic patients in whom symptoms developed as a working adult (especially recent onset), or in whom the severity of asthma has substantially increased. OA should also be considered in any other individuals who have asthma-like symptoms and work in occupations in which they are exposed to asthma-causing agents or who are concerned that their symptoms are work-related.
Patients with possible OA should be asked to provide a thorough medical and occupational/environmental history, with careful documentation of the nature and date of onset of symptoms and diagnosis of asthma, and any potentially causal exposures at that time. Compatibility of the medical history with the clinical presentation of OA described above should be evaluated, especially the temporal pattern of symptoms in relation to work schedule and changes in work exposures. Patterns and changes in patterns of use of asthma medications, and the minimum period of time away from work required for improvement in symptoms should be noted. Prior respiratory diseases, allergies/atopy, smoking and other toxic exposures, and a family history of allergy are pertinent.
Occupational and other environmental exposures to potential asthma-causing agents or processes should be thoroughly explored, with objective documentation of exposures if possible. Suspected exposures should be compared with a comprehensive list of agents reported to cause OA (Harber, Schenker and Balmes 1996; Chan-Yeung and Malo 1994; Bernstein et al. 1993; Rom 1992b), although inability to identify specific agents is not uncommon and induction of asthma by agents not previously described is possible as well. Some illustrative examples are shown in table 3. Occupational history should include details of current and relevant past employment with dates, job titles, tasks and exposures, especially current job and job held at time of onset of symptoms. Other environmental history should include a review of exposures in the home or community that could cause asthma. It is helpful to begin the exposure history in an open-ended way, asking about broad categories of airborne agents: dusts (especially organic dusts of animal, plant or microbial origin), chemicals, pharmaceuticals and irritating or visible gases or fumes. The patient may identify specific agents, work processes or generic categories of agents that have triggered symptoms. Asking the patient to describe step by step the activities and exposures involved in the most recent symptomatic workday can provide useful clues. Materials used by co-workers, or those released in high concentration from a spill or other source, may be relevant. Further information can often be obtained on product name, ingredients and manufacturer name, address and phone number. Specific agents can be identified by calling the manufacturer or through a variety of other sources including textbooks, CD ROM databases, or Poison Control Centers. Since OA is frequently caused by low levels of airborne allergens, workplace industrial hygiene inspections which qualitatively evaluate exposures and control measures are often more helpful than quantitative measurement of air contaminants.
Table 3. Sensitizing agents that can cause occupational asthma
|
Classification |
Sub-groups |
Examples of substances |
Examples of jobs and industries |
|
High-molecular-weight protein antigens |
Animal-derived substances Plant-derived substances |
Laboratory animals, crab/seafood, mites, insects Flour and grain dusts, natural rubber latex gloves, bacterial enzymes, castor bean dust, vegetable gums |
Animal handlers, farming and food processing Bakeries, health care workers, detergent making, food processing |
|
Low-molecular-weight/chemical |
Plasticizers, 2-part paints, adhesives, foams Metals Wood dusts Pharmaceuticals, drugs |
Isocyanates, acid anhydrides, amines Platinum salts, cobalt Cedar (plicatic acid), oak Psyllium, antibiotics |
Auto spray painting, varnishing, woodworking Platinum refineries, metal grinding Sawmill work, carpentry Pharmaceutical manufacturing and packaging |
|
Other chemicals |
Chloramine T, polyvinyl chloride fumes, organophosphate insecticides |
Janitorial work, meat packing |
The clinical history appears to be better for excluding rather than for confirming the diagnosis of OA, and an open-ended history taken by a physician is better than a closed questionnaire. One study compared the results of an open-ended clinical history taken by trained OA specialists with a “gold standard” of specific bronchial challenge testing in 162 patients referred for evaluation of possible OA. The investigators reported that the sensitivity of a clinical history suggestive of OA was 87%, specificity 55%, predictive value positive 63% and predictive value negative 83%. In this group of referred patients, prevalence of asthma and OA were 80% and 46%, respectively (Malo et al. 1991). In other groups of referred patients, predictive values positive of a closed questionnaire ranged from 8 to 52% for a variety of workplace exposures (Bernstein et al. 1993). The applicability of these results to other settings needs to be assessed by the physician.
Physical examination is sometimes helpful, and findings relevant to asthma (e.g., wheezing, nasal polyps, eczematous dermatitis), respiratory irritation or allergy (e.g., rhinitis, conjunctivitis) or other potential causes of symptoms should be noted.
Step 2: Physiological evaluation for reversible airway obstruction and/or non-specific bronchial hyper-responsiveness
If sufficient physiological evidence supporting the diagnosis of asthma (NAEP 1991) is already in the medical record, Step 2 can be skipped. If not, technician-coached spirometry should be performed, preferably post-workshift on a day when the patient is experiencing asthmatic symptoms. If spirometry reveals airway obstruction which reverses with a bronchodilator, this confirms the diagnosis of asthma. In patients without clear evidence of airflow limitation on spirometry, quantitative testing for NBR using methacholine or histamine should be done, the same day if possible. Quantitative testing for NBR in this situation is a key procedure for two reasons. First, it can often identify patients with mild or early stage OA who have the greatest potential for cure but who would be missed if testing stopped with normal spirometry. Second, if NBR is normal in a worker who has ongoing exposure in the workplace environment associated with the symptoms, OA can generally be ruled out without further testing. If abnormal, evaluation can proceed to Step 3 or 4, and the degree of NBR may be useful in monitoring the patient for improvement after diagnostic trial of removal from the suspected causal exposure (Step 5). If spirometry reveals significant airflow limitation that does not improve after inhaled bronchodilator, a re-evaluation after more prolonged trial of therapy, including corticosteroids, should be considered (ATS 1995; NAEP 1991).
Step 3: Immunological assessment, if appropriate
Skin or serological (e.g., RAST) testing can demonstrate immunological sensitization to a specific workplace agent. These immunological tests have been used to confirm the work-relatedness of asthma, and, in some cases, eliminate the need for specific inhalation challenge tests. For example, among psyllium-exposed patients with a clinical history compatible with OA, documented asthma or airway hyper-responsiveness, and evidence of immunological sensitization to psyllium, approximately 80% had OA confirmed on subsequent specific bronchial challenge testing (Malo et al. 1990). In most cases, diagnostic significance of negative immunological tests is less clear. The diagnostic sensitivity of the immunological tests depends critically on whether all the likely causal antigens in the workplace or hapten-protein complexes have been included in the testing. Although the implication of sensitization for an asymptomatic worker is not well defined, analysis of grouped results can be useful in evaluating environmental controls. The utility of immunological evaluation is greatest for agents for which there are standardized in vitro tests or skin-prick reagents, such as platinum salts and detergent enzymes. Unfortunately, most occupational allergens of interest are not currently available commercially. The use of non-commercial solutions in skin-prick testing has on occasions been associated with severe reactions, including anaphylaxis, and thus caution is necessary.
If results of Steps 1 and 2 are compatible with OA, further evaluation should be pursued if possible. The order and extent of further evaluation depends on availability of diagnostic resources, work status of the patient and feasibility of diagnostic trials of removal from and return to work as indicated in Table 7. If further evaluation is not possible, a diagnosis must be based on the information available at this point.
Step 4: Clinical evaluation of asthma at work, or diagnostic trial of return to work
Often the most readily available physiological test of airway obstruction is spirometry. To improve reproducibility, spirometry should be coached by a trained technician. Unfortunately, single-day cross-shift spirometry, performed before and after the workshift, is neither sensitive nor specific in determining work-associated airway obstruction. It is probable that if multiple spirometries are performed each day during and after several workdays, the diagnostic accuracy may be improved, but this has not yet been adequately evaluated.
Due to difficulties with cross-shift spirometry, serial PEF measurement has become an important diagnostic technique for OA. Using an inexpensive portable meter, PEF measurements are recorded every two hours, during waking hours. To improve sensitivity, measurements must be done during a period when the worker is exposed to the suspected causal agents at work and is experiencing a work-related pattern of symptoms. Three repetitions are performed at each time, and measurements are made every day at work and away from work. The measurements should be continued for at least 16 consecutive days (e.g., two five-day work weeks and 3 weekends off) if the patient can safely tolerate continuing to work. PEF measurements are recorded in a diary along with notation of work hours, symptoms, use of bronchodilator medications, and significant exposures. To facilitate interpretation, the diary results should then be plotted graphically. Certain patterns suggest OA, but none are pathognomonic, and interpretation by an experienced reader is often helpful. Advantages of serial PEF testing are low cost and reasonable correlation with results of bronchial challenge testing. Disadvantages include the significant degree of patient cooperation required, inability to definitely confirm that data are accurate, lack of standardized method of interpretation, and the need for some patients to take 1 or 2 consecutive weeks off work to show significant improvement. Portable electronic recording spirometers designed for patient self monitoring, when available, can address some of the disadvantages of serial PEF.
Asthma medications tend to reduce the effect of work exposures on measures of airflow. However, it is not advisable to discontinue medications during airflow monitoring at work. Rather, the patient should be maintained on a constant minimal safe dosage of anti-inflammatory medications throughout the entire diagnostic process, with close monitoring of symptoms and airflow, and the use of short-acting bronchodilators to control symptoms should be noted in the diary.
The failure to observe work-related changes in PEF while a patient is working routine hours does not exclude the diagnosis of OA, since many patients will require more than a two-day weekend to show significant improvement in PEF. In this case, a diagnostic trial of extended removal from work (Step 5) should be considered. If the patient has not yet had quantitative testing for NBR, and does not have a medical contra-indication, it should be done at this time, immediately after at least two weeks of workplace exposure.
Step 5: Clinical evaluation of asthma away from work or diagnostic trial of extended removal from work
This step consists of completion of the serial 2-hourly PEF daily diary for at least 9 consecutive days away from work (e.g., 5 days off work plus weekends before and after). If this record, compared with the serial PEF diary at work, is not sufficient for diagnosing OA, it should be continued for a second consecutive week away from work. After 2 or more weeks away from work, quantitative testing for NBR can be performed and compared to NBR while at work. If serial PEF has not yet been done during at least two weeks at work, then a diagnostic trial of return to work (see Step 4) may be performed, after detailed counselling, and in close contact with the treating physician. Step 5 is often critically important in confirming or excluding the diagnosis of OA, although it may also be the most difficult and expensive step. If an extended removal from work is attempted, it is best to maximize the diagnostic yield and efficiency by including PEF, FEV1, and NBR tests in one comprehensive evaluation. Weekly physician visits for counselling and to review the PEF chart can help to assure complete and accurate results. If, after monitoring the patient for at least two weeks at work and two weeks away from it, the diagnostic evidence is not yet sufficient, Step 6 should be considered next, if available and feasible.
Step 6: Specific bronchial challenge or workplace challenge testing
Specific bronchial challenge testing using an exposure chamber and standardized exposure levels has been labelled the “gold standard” for diagnosis of OA. Advantages include definitive confirmation of OA with ability to identify asthmatic response to sub-irritant levels of specific sensitizing agents, which can then be scrupulously avoided. Of all the diagnostic methods, it is the only one that can reliably distinguish sensitizer-induced asthma from provocation by irritants. Several problems with this approach have included inherent costliness of the procedure, general requirement of close observation or hospitalization for several days, and availability in only very few specialized centres. False negatives may occur if standardized methodology is not available for all suspected agents, if the wrong agents are suspected, or if too long a time has elapsed between last exposure and testing. False positives may result if irritant levels of exposure are inadvertently obtained. For these reasons, specific bronchial challenge testing for OA remains a research procedure in most localities.
Workplace challenge testing involves serial technician-coached spirometry in the workplace, performed at frequent (e.g., hourly) intervals before and during the course of a workday exposure to the suspected causal agents or processes. It may be more sensitive than specific bronchial challenge testing because it involves “real life” exposures, but since airway obstruction may be triggered by irritants as well as sensitizing agents, positive tests do not necessarily indicate sensitization. It also requires cooperation of the employer and much technician time with a mobile spirometer. Both of these procedures carry some risk of precipitating a severe asthmatic attack, and should therefore be done under close supervision of specialists experienced with the procedures.
Treatment and Prevention
Management of OA includes medical and preventive interventions for individual patients, as well as public health measures in workplaces identified as high risk for OA. Medical management is similar to that for non-occupational asthma and is well reviewed elsewhere (NAEP 1991). Medical management alone is rarely adequate to optimally control symptoms, and preventive intervention by control or cessation of exposure is an integral part of the treatment. This process begins with accurate diagnosis and identification of causative exposures and conditions. In sensitizer-induced OA, reducing exposure to the sensitizer does not usually result in complete resolution of symptoms. Severe asthmatic episodes or progressive worsening of the disease may be caused by exposures to very low concentrations of the agent and complete and permanent cessation of exposure is recommended. Timely referral for vocational rehabilitation and job retraining may be a necessary component of treatment for some patients. If complete cessation of exposure is impossible, substantial reduction of exposure accompanied by close medical monitoring and management may be an option, although such reduction in exposure is not always feasible and the long-term safety of this approach has not been tested. As an example, it would be difficult to justify the toxicity of long-term treatment with systemic corticosteroids in order to allow the patient to continue in the same employment. For asthma induced and/or triggered by irritants, dose response may be more predictable, and lowering of irritant exposure levels, accompanied by close medical monitoring, may be less risky and more likely to be effective than for sensitizer-induced OA. If the patient continues to work under modified conditions, medical follow-up should include frequent physician visits with review of the PEF diary, well-planned access to emergency services, and serial spirometry and/or methacholine challenge testing, as appropriate.
Once a particular workplace is suspected to be high risk, due either to occurrence of a sentinel case of OA or use of known asthma-causing agents, public health methods can be very useful. Early recognition and effective treatment and prevention of disability of workers with existing OA, and prevention of new cases, are clear priorities. Identification of specific causal agent(s) and work processes is important. One practical initial approach is a workplace questionnaire survey, evaluating criteria A, B, C, and D1 or D5 in the case definition of OA. This approach can identify individuals for whom further clinical evaluation might be indicated and help identify possible causal agents or circumstances. Evaluation of group results can help decide whether further workplace investigation or intervention is indicated and, if so, provide valuable guidance in targeting future prevention efforts in the most effective and efficient manner. A questionnaire survey is not adequate, however, to establish individual medical diagnoses, since predictive positive values of questionnaires for OA are not high enough. If a greater level of diagnostic certainty is needed, medical screening utilizing diagnostic procedures such as spirometry, quantitative testing for NBR, serial PEF recording, and immunological testing can be considered as well. In known problem workplaces, ongoing surveillance and screening programmes may be helpful. However, differential exclusion of asymptomatic workers with history of atopy or other potential susceptibility factors from workplaces believed to be high risk would result in removal of large numbers of workers to prevent relatively few cases of OA, and is not supported by the current literature.
Control or elimination of causal exposures and avoidance and proper management of spills or episodes of high-level exposures can lead to effective primary prevention of sensitization and OA in co-workers of the sentinel case. The usual exposure control hierarchy of substitution, engineering and administrative controls, and personal protective equipment, as well as education of workers and managers, should be implemented as appropriate. Proactive employers will initiate or participate in some or all of these approaches, but in the event that inadequate preventive action is taken and workers remain at high risk, governmental enforcement agencies may be helpful.
Impairment and Disability
Medical impairment is a functional abnormality resulting from a medical condition. Disability refers to the total effect of the medical impairment on the patient’s life, and is influenced by many non-medical factors such as age and socio-economic status (ATS 1995).
Assessment of medical impairment is done by the physician and may include a calculated impairment index, as well as other clinical considerations. The impairment index is based on (1) degree of airflow limitation after bronchodilator, (2) either degree of reversibility of airflow limitation with bronchodilator or degree of airway hyper-responsiveness on quantitative testing for NBR, and (3) minimum medication required to control asthma. The other major component of the assessment of medical impairment is the physician’s medical judgement of the ability of the patient to work in the workplace environment causing the asthma. For example, a patient with sensitizer-induced OA may have a medical impairment which is highly specific to the agent to which he or she has become sensitized. The worker who experiences symptoms only when exposed to this agent may be able to work in other jobs, but permanently unable to work in the specific job for which she or he has the most training and experience.
Assessment of disability due to asthma (including OA) requires consideration of medical impairment as well as other non-medical factors affecting ability to work and function in everyday life. Disability assessment is initially made by the physician, who should identify all the factors affecting the impact of the impairment on the patient’s life. Many factors such as occupation, educational level, possession of other marketable skills, economic conditions and other social factors may lead to varying levels of disability in individuals with the same level of medical impairment. This information can then be used by administrators to determine disability for purposes of compensation.
Impairment and disability may be classified as temporary or permanent, depending on the likelihood of significant improvement, and whether effective exposure controls are successfully implemented in the workplace. For example, an individual with sensitizer-induced OA is generally considered permanently, totally impaired for any job involving exposure to the causal agent. If the symptoms resolve partially or completely after cessation of exposure, these individuals may be classified with less or no impairment for other jobs. Often this is considered permanent partial impairment/disability, but terminology may vary. An individual with asthma which is triggered in a dose-dependent fashion by irritants in the workplace would be considered to have temporary impairment while symptomatic, and less or no impairment if adequate exposure controls are installed and are effective in reducing or eliminating symptoms. If effective exposure controls are not implemented, the same individual might have to be considered permanently impaired to work in that job, with recommendation for medical removal. If necessary, repeated assessment for long-term impairment/disability may be carried out two years after the exposure is reduced or terminated, when improvement of OA would be expected to have plateaued. If the patient continues to work, medical monitoring should be ongoing and reassessment of impairment/disability should be repeated as needed.
Workers who become disabled by OA or WAA may qualify for financial compensation for medical expenses and/or lost wages. In addition to directly reducing the financial impact of the disability on individual workers and their families, compensation may be necessary to provide proper medical treatment, initiate preventive intervention and obtain vocational rehabilitation. The worker’s and physician’s understanding of specific medico-legal issues may be important to ensuring that the diagnostic evaluation meets local requirements and does not result in compromise of the rights of the affected worker.
Although discussions of cost savings frequently focus on the inadequacy of compensation systems, genuinely reducing the financial and public health burden placed on society by OA and WAA will depend not only on improvements in compensation systems but, more importantly, on effectiveness of the systems deployed to identify and rectify, or prevent entirely, workplace exposures that are causing onset of new cases of asthma.
Conclusions
OA has become the most prevalent occupational respiratory disease in many countries. It is more common than generally recognized, can be severe and disabling, and is generally preventable. Early recognition and effective preventive interventions can substantially reduce the risk of permanent disability and the high human and financial costs associated with chronic asthma. For many reasons, OA merits more widespread attention among clinicians, health and safety specialists, researchers, health policy makers, industrial hygienists, and others interested in prevention of work-related diseases.
Diseases Caused by Respiratory Irritants and Toxic Chemicals
The presence of respiratory irritants in the workplace can be unpleasant and distracting, leading to poor morale and decreased productivity. Certain exposures are dangerous, even lethal. In either extreme, the problem of respiratory irritants and inhaled toxic chemicals is common; many workers face a daily threat of exposure. These compounds cause harm by a variety of different mechanisms, and the extent of injury can vary widely, depending on the degree of exposure and on the biochemical properties of the inhalant. However, they all have the characteristic of nonspecificity; that is, above a certain level of exposure virtually all persons experience a threat to their health.
There are other inhaled substances that cause only susceptible individuals to develop respiratory problems; such complaints are most appropriately approached as diseases of allergic and immunological origin. Certain compounds, such as isocyanates, acid anhydrides and epoxy resins, can act not only as non-specific irritants in high concentrations, but can also predispose certain subjects to allergic sensitization. These compounds provoke respiratory symptoms in sensitized individuals at very low concentrations.
Respiratory irritants include substances that cause inflammation of the airways after they are inhaled. Damage may occur in the upper and lower airways. More dangerous is acute inflammation of the pulmonary parenchyma, as in chemical pneumonitis or non-cardiogenic pulmonary oedema. Compounds that can cause parenchymal damage are considered toxic chemicals. Many inhaled toxic chemicals also act as respiratory irritants, warning us of their danger with their noxious odour and symptoms of nose and throat irritation and cough. Most respiratory irritants are also toxic to the lung parenchyma if inhaled in sufficient amount.
Many inhaled substances have systemic toxic effects after being absorbed by inhalation. Inflammatory effects on the lung may be absent, as in the case of lead, carbon monoxide or hydrogen cyanide. Minimal lung inflammation is normally seen in the inhalation fevers (e.g., organic dust toxic syndrome, metal fume fever and polymer fume fever). Severe lung and distal organ damage occurs with significant exposure to toxins such as cadmium and mercury.
The physical properties of inhaled substances predict the site of deposition; irritants will produce symptoms at these sites. Large particles (10 to 20mm) deposit in the nose and upper airways, smaller particles (5 to 10mm) deposit in the trachea and bronchi, and particles less than 5mm in size may reach the alveoli. Particles less than 0.5mm are so small they behave like gases. Toxic gases deposit according to their solubility. A water-soluble gas will be adsorbed by the moist mucosa of the upper airway; less soluble gases will deposit more randomly throughout the respiratory tract.
Respiratory Irritants
Respiratory irritants cause non-specific inflammation of the lung after being inhaled. These substances, their sources of exposure, physical and other properties, and effects on the victim are outlined in Table 1. Irritant gases tend to be more water soluble than gases more toxic to the lung parenchyma. Toxic fumes are more dangerous when they have a high irritant threshold; that is, there is little warning that the fume is being inhaled because there is little irritation.
Table 1. Summary of respiratory irritants
|
Chemical |
Sources of exposure |
Important properties |
Injury produced |
Dangerous exposure level under 15 min (PPM) |
|
Acetaldehyde |
Plastics, synthetic rubber industry, combustion products |
High vapour pressure; high water solubility |
Upper airway injury; rarely causes delayed pulmonary oedema |
|
|
Acetic acid, organic acids |
Chemical industry, electronics, combustion products |
Water soluble |
Ocular and upper airway injury |
|
|
Acid anhydrides |
Chemicals, paints, and plastics industries; components of epoxy resins |
Water soluble, highly reactive, may cause allergic sensitization |
Ocular, upper airway injury, bronchospasm; pulmonary haemorrhage after massive exposure |
|
|
Acrolein |
Plastics, textiles, pharmaceutical manufacturing, combustion products |
High vapour pressure, intermediate water solubility, extremely irritating |
Diffuse airway and parenchymal injury |
|
|
Ammonia |
Fertilizers, animal feeds, chemicals, and pharmaceuticals manufacturing |
Alkaline gas, very high water solubility |
Primarily ocular and upper airway burn; massive exposure may cause bronchiectasis |
500 |
|
Antimony trichloride, antimony penta-chloride |
Alloys, organic catalysts |
Poorly soluble, injury likely due to halide ion |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
|
Beryllium |
Alloys (with copper), ceramics; electronics, aerospace and nuclear reactor equipment |
Irritant metal, also acts as an antigen to promote a long-term granulomatous response |
Acute upper airway injury, tracheobronchitis, chemical pneumonitis |
25 μg/m3 |
|
Boranes (diborane) |
Aircraft fuel, fungicide manufacturing |
Water soluble gas |
Upper airway injury, pneumonitis with massive exposure |
|
|
Hydrogen bromide |
Petroleum refining |
Upper airway injury, pneumonitis with massive exposure |
||
|
Methyl bromide |
Refrigeration, produce fumigation |
Moderately soluble gas |
Upper and lower airway injury, pneumonitis, CNS depression and seizures |
|
|
Cadmium |
Alloys with Zn and Pb, electroplating, batteries, insecticides |
Acute and chronic respiratory effects |
Tracheobronchitis, pulmonary oedema (often delayed onset over 24–48 hours); chronic low level exposure leads to inflammatory changes and emphysema |
100 |
|
Calcium oxide, calcium hydroxide |
Lime, photography, tanning, insecticides |
Moderately caustic, very high doses required for toxicity |
Upper and lower airway inflammation, pneumonitis |
|
|
Chlorine |
Bleaching, formation of chlorinated compounds, household cleaners |
Intermediate water solubilty |
Upper and lower airway inflammation, pneumonitis and non-cardiogenic pulmonary oedema |
5–10 |
|
Chloroacetophenone |
Crowd control agent, “tear gas” |
Irritant qualities are used to incapacitate; alkylating agent |
Ocular and upper airway inflammation, lower airway and parenchymal injury with masssive exposure |
1–10 |
|
o-Chlorobenzomalo- nitrile |
Crowd control agent, “tear gas” |
Irritant qualities are used to incapacitate |
Ocular and upper airway inflammation, lower airway injury with massive exposure |
|
|
Chloromethyl ethers |
Solvents, used in manufacture of other organic compounds |
Upper and lower airway irritation, also a respiratory tract carcinogen |
||
|
Chloropicrin |
Chemical manufacturing, fumigant component |
Former First World War gas |
Upper and lower airway inflammation |
15 |
|
Chromic acid (Cr(IV)) |
Welding, plating |
Water soluble irritant, allergic sensitizer |
Nasal inflammation and ulceration, rhinitis, pneumonitis with massive exposure |
|
|
Cobalt |
High temperature alloys, permanent magnets, hard metal tools (with tungsten carbide) |
Non-specific irritant, also allergic sensitizer |
Acute bronchospasm and/or pneumonitis; chronic exposure can cause lung fibrosis |
|
|
Formaldehyde |
Manufacture of foam insulation, plywood, textiles, paper, fertilizers, resins; embalming agents; combustion products |
Highly water soluble, rapidly metabolized; primarily acts via sensory nerve stimulation; sensitization reported |
Ocular and upper airway irritation; bronchospasm in severe exposure; contact dermatitis in sensitized persons |
3 |
|
Hydrochloric acid |
Metal refining, rubber manufacturing, organic compound manufacture, photographic materials |
Highly water soluble |
Ocular and upper airway inflammation, lower airway inflammation only with massive exposure |
100 |
|
Hydrofluoric acid |
Chemical catalyst, pesticides, bleaching, welding, etching |
Highly water soluble, powerful and rapid oxidant, lowers serum calcium in massive exposure |
Ocular and upper airway inflammation, tracheobronchitis and pneumonitis with massive exposure |
20 |
|
Isocyanates |
Polyurethane production; paints; herbicide and insecticide products; laminating, furniture, enamelling, resin work |
Low molecular weight organic compounds, irritants, cause sensitization in susceptible persons |
Ocular, upper and lower inflammation; asthma, hypersensitivity pneumonitis in sensitized persons |
0.1 |
|
Lithium hydride |
Alloys, ceramics, electronics, chemical catalysts |
Low solubility, highly reactive |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
|
Mercury |
Electrolysis, ore and amalgam extraction, electronics manufacture |
No respiratory symptoms with low level, chronic exposure |
Ocular and respiratory tract inflammation, pneumonitis, CNS, kidney and systemic effects |
1.1 mg/m3 |
|
Nickel carbonyl |
Nickel refining, electroplating, chemical reagents |
Potent toxin |
Lower respiratory irritation, pneumonitis, delayed systemic toxic effects |
8 μg/m3 |
|
Nitrogen dioxide |
Silos after new grain storage, fertilizer making, arc welding, combustion products |
Low water solubility, brown gas at high concentration |
Ocular and upper airway inflammation, non-cardiogenic pulmonary oedema, delayed onset bronchiolitis |
50 |
|
Nitrogen mustards; sulphur mustards |
Military gases |
Causes severe injury, vesicant properties |
Ocular, upper and lower airway inflammation, pneumonitis |
20mg/m3 (N) 1 mg/m3 (S) |
|
Osmium tetroxide |
Copper refining, alloy with iridium, catalyst for steroid synthesis and ammonia formation |
Metallic osmium is inert, tetraoxide forms when heated in air |
Severe ocular and upper airway irritation; transient renal damage |
1 mg/m3 |
|
Ozone |
Arc welding, copy machines, paper bleaching |
Sweet smelling gas, moderate water solubility |
Upper and lower airway inflammation; asthmatics more susceptible |
1 |
|
Phosgene |
Pesticide and other chemical manufacture, arc welding, paint removal |
Poorly water soluble, does not irritate airways in low doses |
Upper airway inflammation and pneumonitis; delayed pulmonary oedema in low doses |
2 |
|
Phosphoric sulphides |
Production of insecticides, ignition compounds, matches |
Ocular and upper airway inflammation |
||
|
Phosphoric chlorides |
Manufacture of chlorinated organic compounds, dyes, gasoline additives |
Form phosphoric acid and hydrochloric acid on contact with mucosal surfaces |
Ocular and upper airway inflammation |
10 mg/m3 |
|
Selenium dioxide |
Copper or nickel smelting, heating of selenium alloys |
Strong vessicant, forms selenious acid (H2SeO3) on mucosal surfaces |
Ocular and upper airway inflammation, pulmonary oedema in massive exposure |
|
|
Hydrogen selenide |
Copper refining, sulphuric acid production |
Water soluble; exposure to selenium compounds gives rise to garlic odour breath |
Ocular and upper airway inflammation, delayed pulmonary oedema |
|
|
Styrene |
Manufacture of polystyrene and resins, polymers |
Highly irritating |
Ocular, upper and lower airway inflammation, neurological impairments |
600 |
|
Sulphur dioxide |
Petroleum refining, pulp mills, refrigeration plants, manufacturing of sodium sulphite |
Highly water soluble gas |
Upper airway inflammation, bronchoconstriction, pneumonitis on massive exposure |
100 |
|
Titanium tetrachloride |
Dyes, pigments, sky writing |
Chloride ions form HCl on mucosa |
Upper airway injury |
|
|
Uranium hexafluoride |
Metal coat removers, floor sealants, spray paints |
Toxicity likely from chloride ions |
Upper and lower airway injury, bronchospasm, pneumonitis |
|
|
Vanadium pentoxide |
Cleaning oil tanks, metallurgy |
Ocular, upper and lower airway symptoms |
70 |
|
|
Zinc chloride |
Smoke grenades, artillery |
More severe than zinc oxide exposure |
Upper and lower airway irritation, fever, delayed onset pneumonitis |
200 |
|
Zirconium tetrachloride |
Pigments, catalysts |
Chloride ion toxicity |
Upper and lower airway irritation, pneumonitis |
This condition is thought to result from persistent inflammation with reduction of epithelial cell layer permeability or reduced conductance threshold for subepithelial nerve endings.Adapted from Sheppard 1988; Graham 1994; Rom 1992; Blanc and Schwartz 1994; Nemery 1990; Skornik 1988.
The nature and extent of the reaction to an irritant depends on the physical properties of the gas or aerosol, the concentration and time of exposure, and on other variables as well, such as temperature, humidity and the presence of pathogens or other gases (Man and Hulbert 1988). Host factors such as age (Cabral-Anderson, Evans and Freeman 1977; Evans, Cabral-Anderson and Freeman 1977), prior exposure (Tyler, Tyler and Last 1988), level of antioxidants (McMillan and Boyd 1982) and presence of infection may play a role in determining the pathological changes seen. This wide range of factors has made it difficult to study the pathogenic effects of respiratory irritants in a systematic way.
The best understood irritants are those which inflict oxidative injury. The majority of inhaled irritants, including the major pollutants, act by oxidation or give rise to compounds that act in this way. Most metal fumes are actually oxides of the heated metal; these oxides cause oxidative injury. Oxidants damage cells primarily by lipid peroxidation, and there may be other mechanisms. On a cellular level, there is initially a fairly specific loss of ciliated cells of the airway epithelium and of Type I alveolar epithelial cells, with subsequent violation of the tight junction interface between epithelial cells (Man and Hulbert 1988; Gordon, Salano and Kleinerman 1986; Stephens et al. 1974). This leads to subepithelial and submucosal damage, with stimulation of smooth muscle and parasympathetic sensory afferent nerve endings causing bronchoconstriction (Holgate, Beasley and Twentyman 1987; Boucher 1981). An inflammatory response follows (Hogg 1981), and the neutrophils and eosinophils release mediators that cause further oxidative injury (Castleman et al. 1980). Type II pneumocytes and cuboidal cells act as stem cells for repair (Keenan, Combs and McDowell 1982; Keenan, Wilson and McDowell 1983).
Other mechanisms of lung injury eventually involve the oxidative pathway of cellular damage, particularly after damage to the protective epithelial cell layer has occurred and an inflammatory response has been elicited. The most commonly described mechanisms are outlined in table 2.
Table 2. Mechanisms of lung injury by inhaled substances
|
Mechanism of injury |
Example compounds |
Damage that occurs |
|
Oxidation |
Ozone, nitrogen dioxide, sulphur dioxide, chlorine, oxides |
Patchy airway epithelial damage, with increased permeability and exposure of nerve fibre endings; loss of cilia from ciliated cells; necrosis of type I pneumocytes; free radical formation and subsequent protein binding and lipid peroxidation |
|
Acid formation |
Sulphur dioxide, chlorine, halides |
Gas dissolves in water to form acid that damages epithelial cells via oxidation; action mainly on upper airway |
|
Alkali formation |
Ammonia, calcium oxide, hydroxides |
Gas dissolves in water to form alkaline solution that may cause tissue liquefaction; predominant upper airway damage, lower airway in heavy exposures |
|
Protein binding |
Formaldehyde |
Reactions with amino acids lead to toxic intermediates with damage to the epithelial cell layer |
|
Afferent nerve stimulation |
Ammonia, formaldehyde |
Direct nerve ending stimulation provokes symptoms |
|
Antigenicity |
Platinum, acid anhydrides |
Low molecular weight molecules serve as haptens in sensitized persons |
|
Stimulation of host inflammatory response |
Copper and zinc oxides, lipoproteins |
Stimulation of cytokines and inflammatory mediators without apparent direct cellular damage |
|
Free radical formation |
Paraquat |
Promotion of formation or retardation of clearance of superoxide radicals, leading to lipid peroxidation and oxidative damage |
|
Delayed particle clearance |
Any prolonged inhalation of mineral dust |
Overwhelming of mucociliary escalators and alveolar macrophage systems with particles, leading to a non-specific inflammatory response |
Workers exposed to low levels of respiratory irritants may have subclinical symptoms traceable to mucous membrane irritation, such as watery eyes, sore throat, runny nose and cough. With significant exposure, the added feeling of shortness of breath will often prompt medical attention. It is important to secure a good medical history in order to determine the likely composition of the exposure, the quantity of exposure, and the period of time during which the exposure took place. Signs of laryngeal oedema, including hoarseness and stridor, should be sought, and the lungs should be examined for signs of lower airway or parenchymal involvement. Assessment of the airway and lung function, together with chest radiography, are important in short-term management. Laryngoscopy may be indicated to evaluate the airway.
If the airway is threatened, the patient should undergo intubation and supportive care. Patients with signs of laryngeal oedema should be observed for at least 12 hours to insure that the process is self-limited. Bronchospasm should be treated with b-agonists and, if refractory, intravenous corticosteroids. Irritated oral and ocular mucosa should be thoroughly irrigated. Patients with crackles on examination or chest radiograph abnormalities should be hospitalized for observation in view of the possibility of pneumonitis or pulmonary oedema. Such patients are at risk of bacterial superinfection; nevertheless, no benefit has been demonstrated by using prophylactic antibiotics.
The overwhelming majority of patients who survive the initial insult recover fully from irritant exposures. The chances for long-term sequelae are more likely with greater initial injury. The term reactive airway dysfunction syndrome (RADS) has been applied to the persistence of asthma-like symptoms following acute exposure to respiratory irritants (Brooks, Weiss and Bernstein 1985).
High-level exposures to alkalis and acids can cause upper and lower respiratory tract burns that lead to chronic disease. Ammonia is known to cause bronchiectasis (Kass et al. 1972); chlorine gas (which becomes HCl in the mucosa) is reported to cause obstructive lung disease (Donelly and Fitzgerald 1990; Das and Blanc 1993). Chronic low-level exposures to irritants may cause continued ocular and upper airway symptoms (Korn, Dockery and Speizer 1987), but deterioration of lung function has not been conclusively documented. Studies of the effects of chronic low-level irritants on airway function are hampered by a lack of long-term follow-up, confounding by cigarette smoking, the “healthy worker effect,” and the minimal, if any, actual clinical effect (Brooks and Kalica 1987).
After a patient recovers from the initial injury, regular follow-up by a physician is needed. Clearly, there should be an effort to investigate the workplace and evaluate respiratory precautions, ventilation and containment of the culprit irritants.
Toxic Chemicals
Chemicals toxic to the lung include most of the respiratory irritants given enough high exposure, but there are many chemicals that cause significant parenchymal lung injury despite possessing low to moderate irritant properties. These compounds work their effects by mechanisms reviewed in Table 3 and discussed above. Pulmonary toxins tend to be less water soluble than upper airway irritants. Examples of lung toxins and their sources of exposure are reviewed in table 3.
Table 3. Compounds capable of lung toxicity after low to moderate exposure
|
Compound |
Sources of exposure |
Toxicity |
|
Acrolein |
Plastics, textiles, pharmaceutical manufacturing, combustion products |
Diffuse airway and parenchymal injury |
|
Antimony trichloride; antimony |
Alloys, organic catalysts |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
Cadmium |
Alloys with zinc and lead, electroplating, batteries, insecticides |
Tracheobronchitis, pulmonary oedema (often delayed onset over 24–48 hours), kidney damage: tubule proteinuria |
|
Chloropicrin |
Chemical manufacturing, fumigant components |
Upper and lower airway inflammation |
|
Chlorine |
Bleaching, formation of chlorinated compounds, household cleaners |
Upper and lower airway inflammation, pneumonitis and non-cardiogenic pulmonary oedema |
|
Hydrogen sulphide |
Natural gas wells, mines, manure |
Ocular, upper and lower airway irritation, delayed pulmonary oedema, asphyxiation from systemic tissue hypoxia |
|
Lithium hydride |
Alloys, ceramics, electronics, chemical catalysts |
Pneumonitis, non-cardiogenic pulmonary oedema |
|
Methyl isocyanate |
Pesticide synthesis |
Upper and lower respiratory tract irritation, pulmonary oedema |
|
Mercury |
Electrolysis, ore and amalgam extraction, electronics manufacture |
Ocular and respiratory tract inflammation, pneumonitis, CNS, kidney and systemic effects |
|
Nickel carbonyl |
Nickel refining, electroplating, chemical reagents |
Lower respiratory irritation, pneumonitis, delayed systemic toxic effects |
|
Nitrogen dioxide |
Silos after new grain storage, fertilizer making, arc welding; combustion products |
Ocular and upper airway inflammation, non-cardiogenic pulmonary oedema, delayed onset bronchiolitis |
|
Nitrogen mustards, sulphur |
Military agents, vesicants |
Ocular and respiratory tract inflammation, pneumonitis |
|
Paraquat |
Herbicides (ingested) |
Selective damage to type-2 pneumocytes leading to RADS, pulmonary fibrosis; renal failure, GI irritation |
|
Phosgene |
Pesticide and other chemical manufacture, arc welding, paint removal |
Upper airway inflammation and pneumonitis; delayed pulmonary oedema in low doses |
|
Zinc chloride |
Smoke grenades, artillery |
Upper and lower airway irritation, fever, delayed onset pneumonitis |
One group of inhalable toxins are termed asphyxiants. When present in high enough concentrations, the asphyxiants, carbon dioxide, methane and nitrogen, displace oxygen and in effect suffocate the victim. Hydrogen cyanide, carbon monoxide and hydrogen sulphide act by inhibiting cellular respiration despite adequate delivery of oxygen to the lung. Non-asphyxiant inhaled toxins damage target organs, causing a wide variety of health problems and mortality.
The medical management of inhaled lung toxins is similar to the management of respiratory irritants. These toxins often do not elicit their peak clinical effect for several hours after exposure; overnight monitoring may be indicated for compounds known to cause delayed onset pulmonary oedema. Since the therapy of systemic toxins is beyond the scope of this chapter, the reader is referred to discussions of the individual toxins elsewhere in this Encyclopaedia and in further texts on the subject (Goldfrank et al. 1990; Ellenhorn and Barceloux 1988).
Inhalation Fevers
Certain inhalation exposures occurring in a variety of different occupational settings may result in debilitating flu-like illnesses lasting a few hours. These are collectively referred to as inhalation fevers. Despite the severity of the symptoms, the toxicity seems to be self-limited in most cases, and there are few data to suggest long-term sequelae. Massive exposure to inciting compounds can cause a more severe reaction involving pneumonitis and pulmonary oedema; these uncommon cases are considered more complicated than simple inhalation fever.
The inhalation fevers have in common the feature of nonspecificity: the syndrome can be produced in nearly anyone, given adequate exposure to the inciting agent. Sensitization is not required, and no previous exposure is necessary. Some of the syndromes exhibit the phenomenon of tolerance; that is, with regular repeated exposure the symptoms do not occur. This effect is thought to be related to an increased activity of clearance mechanisms, but has not been adequately studied.
Organic Dust Toxic Syndrome
Organic dust toxic syndrome (ODTS) is a broad term denoting the self-limited flu-like symptoms that occur following heavy exposure to organic dusts. The syndrome encompasses a wide range of acute febrile illnesses that have names derived from the specific tasks that lead to dust exposure. Symptoms occur only after a massive exposure to organic dust, and most individuals so exposed will develop the syndrome.
Organic dust toxic syndrome has previously been called pulmonary mycotoxicosis, owing to its putative aetiology in the action of mould spores and actinomycetes. With some patients, one can culture species of Aspergillus, Penicillium, and mesophilic and thermophilic actinomycetes (Emmanuel, Marx and Ault 1975; Emmanuel, Marx and Ault 1989). More recently, bacterial endotoxins have been proposed to play at least as large a role. The syndrome has been provoked experimentally by inhalation of endotoxin derived from Enterobacter agglomerans, a major component of organic dust (Rylander, Bake and Fischer 1989). Endotoxin levels have been measured in the farm environment, with levels ranging from 0.01 to 100μg/m3. Many samples had a level greater than 0.2μg/m3, which is the level where clinical effects are known to occur (May, Stallones and Darrow 1989). There is speculation that cytokines, such as IL-1, may mediate the systemic effects, given what is already known about the release of IL-1 from alveolar macrophages in the presence of endotoxin (Richerson 1990). Allergic mechanisms are unlikely given the lack of need for sensitization and the requirement for high dust exposure.
Clinically, the patient will usually present symptoms 2 to 8 hours after exposure to (usually mouldy) grain, hay, cotton, flax, hemp or wood chips, or upon manipulation of pigs (Do Pico 1992). Often symptoms begin with eye and mucous membrane irritation with dry cough, progressing to fever, and malaise, chest tightness, myalgias and headache. The patient appears ill but otherwise normal upon physical examination. Leukocytosis frequently occurs, with levels as high as 25,000 white blood corpuscles (WBC)/mm3. The chest radiograph is almost always normal. Spirometry may reveal a modest obstructive defect. In cases where fibre optic bronchoscopy was performed and bronchial washings were obtained, an elevation of leukocytes was found in the lavage fluid. The percentage of neutrophils was significantly higher than normal (Emmanuel, Marx and Ault 1989; Lecours, Laviolette and Cormier 1986). Bronchoscopy 1 to 4 weeks after the event shows a persistently high cellularity, predominantly lymphocytes.
Depending on the nature of the exposure, the differential diagnosis may include toxic gas (such as nitrogen dioxide or ammonia) exposure, particularly if the episode occurred in a silo. Hypersensitivity pneumonitis should be considered, especially if there are significant chest radiograph or pulmonary function test abnormalities. The distinction between hypersensitivity pneumonitis (HP) and ODTS is important: HP will require strict exposure avoidance and has a worse prognosis, whereas ODTS has a benign and self-limited course. ODTS is also distinguished from HP because it occurs more frequently, requires higher levels of dust exposure, does not induce the release of serum precipitating antibodies, and (initially) does not give rise to the lymphocytic alveolitis that is characteristic of HP.
Cases are managed with antipyretics. A role for steroids has not been advocated given the self-limited nature of the illness. Patients should be educated about massive exposure avoidance. The long-term effect of repeated occurrences is thought to be negligible; however, this question has not been adequately studied.
Metal Fume Fever
Metal fume fever (MFF) is another self-limited, flu-like illness that develops after inhalation exposure, in this instance to metal fumes. The syndrome most commonly develops after zinc oxide inhalation, as occurs in brass foundries, and in smelting or welding galvanized metal. Oxides of copper and iron also cause MFF, and vapours of aluminium, arsenic, cadmium, mercury, cobalt, chromium, silver, manganese, selenium and tin have been occasionally implicated (Rose 1992). Workers develop tachyphalaxis; that is, symptoms appear only when the exposure occurs after several days without exposure, not when there are regular repeated exposures. An eight-hour TLV of 5 mg/m3 for zinc oxide has been established by the US Occupational Safety and Health Administration (OSHA), but symptoms have been elicited experimentally after a two-hour exposure at this concentration (Gordon et al. 1992).
The pathogenesis of MFF remains unclear. The reproducible onset of symptoms regardless of the individual exposed argues against a specific immune or allergic sensitization. The lack of symptoms associated with histamine release (flushing, itching, wheezing, hives) also militates against the likelihood of an allergic mechanism. Paul Blanc and co-workers have developed a model implicating cytokine release (Blanc et al. 1991; Blanc et al.1993). They measured the levels of tumour necrosis factor (TNF), and of the interleukins IL-1, IL-4, IL-6 and IL-8 in the fluid lavaged from the lungs of 23 volunteers experimentally exposed to zinc oxide fumes (Blanc et al. 1993). The volunteers developed elevated levels of TNF in their bronchoalveolar lavage (BAL) fluid 3 hours after exposure. Twenty hours later, high BAL fluid levels of IL-8 (a potent neutrophil attractant) and an impressive neutrophilic alveolitis were observed. TNF, a cytokine capable of causing fever and stimulating immune cells, has been shown to be released from monocytes in culture that are exposed to zinc (Scuderi 1990). Accordingly, the presence of increased TNF in the lung accounts for the onset of symptoms observed in MFF. TNF is known to stimulate the release of both IL-6 and IL-8, in a time period that correlated with the peaks of the cytokines in these volunteers’ BAL fluid. The recruitment of these cytokines may account for the ensuing neutrophil alveolitis and flu-like symptoms that characterize MFF. Why the alveolitis resolves so quickly remains a mystery.
Symptoms begin 3 to 10 hours after exposure. Initially, there may be a sweet metallic taste in the mouth, accompanied by a worsening dry cough and shortness of breath. Fever and shaking chills often develop, and the worker feels ill. The physical examination is otherwise unremarkable. Laboratory evaluation shows a leukocytosis and a normal chest radiograph. Pulmonary function studies may show a slightly reduced FEF25-75 and DLCO levels (Nemery 1990; Rose 1992).
With a good history the diagnosis is readily established and the worker can be treated symptomatically with antipyretics. Symptoms and clinical abnormalities resolve within 24 to 48 hours. Otherwise, bacterial and viral aetiologies of the symptoms must be considered. In cases of extreme exposure, or exposures involving contamination by toxins such as zinc chloride, cadmium or mercury, MFF may be a harbinger of a clinical chemical pneumonitis that will evolve over the next 2 days (Blount 1990). Such cases can exhibit diffuse infiltrates on a chest radiograph and signs of pulmonary oedema and respiratory failure. While this possibility should be considered in the initial evaluation of an exposed patient, such a fulminant course is unusual and not characteristic of uncomplicated MFF.
MFF does not require a specific sensitivity of the individual for the metal fumes; rather, it indicates inadequate environmental control. The exposure problem should be addressed to prevent recurrent symptoms. Although the syndrome is considered benign, the long-term effects of repeated bouts of MFF have not been adequately investigated.
Polymer Fume Fever
Polymer fume fever is a self-limited febrile illness similar to MFF, but caused by inhaled pyrolysis products of fluoropolymers, including polytetrafluoroethane (PTFE; trade names Teflon, Fluon, Halon). PTFE is widely used for its lubricant, thermal stability and electrical insulative properties. It is harmless unless heated above 30°C, when it starts to release degradation products (Shusterman 1993). This situation occurs when welding materials coated with PTFE, heating PTFE with a tool edge during high speed machining, operating moulding or extruding machines (Rose 1992) and rarely during endotracheal laser surgery (Rom 1992a).
A common cause of polymer fume fever was elicited after a period of classic public health detective work in the early 1970s (Wegman and Peters 1974; Kuntz and McCord 1974). Textile workers were developing self-limited febrile illnesses with exposures to formaldehyde, ammonia and nylon fibre; they did not have exposure to fluoropolymer fumes but handled crushed polymer. After finding that exposure levels of the other possible aetiological agents were within acceptable limits, the fluoropolymer work was examined more closely. As it turned out, only cigarette smokers working with the fluoropolymer were symptomatic. It was hypothesized that the cigarettes were being contaminated with fluoropolymer on the worker’s hands, then the product was combusted on the cigarette when it was smoked, exposing the worker to toxic fumes. After banning cigarette smoking in the workplace and setting strict handwashing rules, no further illnesses were reported (Wegman and Peters 1974). Since then, this phenomenon has been reported after working with waterproofing compounds, mould-release compounds (Albrecht and Bryant 1987) and after using certain kinds of ski wax (Strom and Alexandersen 1990).
The pathogenesis of polymer fume fever is not known. It is thought to be similar to the other inhalation fevers owing to its similar presentation and apparently non-specific immune response. There have been no human experimental studies; however, rats and birds both develop severe alveolar epithelial damage on exposure to PTFE pyrolysis products (Wells, Slocombe and Trapp 1982; Blandford et al. 1975). Accurate measurement of pulmonary function or BAL fluid changes has not been done.
Symptoms appear several hours after exposure, and a tolerance or tachyphalaxis effect is not there as seen in MFF. Weakness and myalgias are followed by fever and chills. Often there is chest tightness and cough. Physical examination is usually otherwise normal. Leukocytosis is often seen, and the chest radiograph is usually normal. Symptoms resolve spontaneously in 12 to 48 hours. There have been a few cases of persons developing pulmonary oedema after exposure; in general, PTFE fumes are thought to be more toxic than zinc or copper fumes in causing MFF (Shusterman 1993; Brubaker 1977). Chronic airways dysfunction has been reported in persons who have had multiple episodes of polymer fume fever (Williams, Atkinson and Patchefsky 1974).
The diagnosis of polymer fume fever requires a careful history with high clinical suspicion. After ascertaining the source of the PTFE pyrolysis products, efforts must be made to prevent further exposure. Mandatory handwashing rules and the elimination of smoking in the workplace has effectively eliminated cases related to contaminated cigarettes. Workers who have had multiple episodes of polymer fume fever or associated pulmonary oedema should have long-term medical follow-up.
Summary Worklife Exposure Measures
Researchers are fortunate when they have at their disposal a detailed chronology of the worklife experience of workers that provides an historic review of jobs they have held over time. For these workers a job exposure matrix can then be set up that allows each and every job change that a worker has gone through to be associated with specific exposure information.
Detailed exposure histories must be summarized for analysis purposes in order to determine whether patterns are evident that could be related to health and safety issues in the workplace. We can visualize a list of, say, 20 job changes that a worker had experienced in his or her working lifetime. There are then several alternative ways in which the exposure details (for each of the 20 job changes in this example) can be summarized, taking duration and/or concentration/dose/grade of exposure into account.
It is important to note, however, that different conclusions from a study could be reached depending on the method selected (Suarez-Almazor et al. 1992). An example of five summary worklife exposure measures is shown in table 1.
Table 1. Formulae and dimensions or units of the five selected summary measures of worklife exposure
|
Exposure measure |
Formula |
Dimensions/Units |
|
Cumulative exposure index (CEI) |
Σ (grade x time exposed) |
grade and time |
|
Mean grade (MG) |
Σ (grade x time exposed)/total time exposed |
grade |
|
Highest grade ever (HG) |
highest grade to which exposed for ≥ 7 days |
grade |
|
Time-weighted average (TWA) grade |
Σ (grade x time exposed)/total time employed |
grade |
|
Total time exposed (TTE) |
Σ time exposed |
time |
Adapted from Suarez-Almazor et al. 1992.
Cumulative exposure index. The cumulative exposure index (CEI) is equivalent to “dose” in toxicological studies and represents the sum, over a working lifetime, of the products of exposure grade and exposure duration for each successive job title. It includes time in its units.
Mean grade. The mean grade (MG) cumulates the products of exposure grade and exposure duration for each successive job title (i.e., the CEI) and divides by the total time exposed at any grade greater than zero. MG is independent of time in its units; the summary measure for a person exposed for a long period at a high concentration will be similar to that for a person exposed for a short period at a high concentration. Within any matched set in a case-control design, MG is an average grade of exposure per unit of time exposed. It is an average grade for the time actually exposed to the agent under consideration.
Highest grade ever. The highest grade ever (HG) is determined from scanning the work history for the highest grade assignment in the period of observation to which the worker was exposed for at least seven days. The HG could misrepresent a person’s worklife exposure because, by its very formulation, it is based on a maximizing rather than on an averaging procedure and is therefore independent of duration of exposure in its units.
Time-weighted average grade. The time-weighted average (TWA) grade is the cumulative exposure index (CEI) divided by the total time employed. Within any matched set in a case-control design, the TWA grade averages over total time employed. It differs from MG, which averages only over the total time actually exposed. Thus, TWA grade can be viewed as an average exposure per unit of time in the full term of employment regardless of exposure per se.
Total time exposed. The total time exposed (TTE) accumulates all time periods associated with exposure in units of time. TTE has appeal for its simplicity. However, it is well accepted that health effects must be related not only to duration of chemical exposure, but also to the intensity of that exposure (i.e., the concentration or grade).
Clearly, the utility of a summary exposure measure is determined by the respective weight it attributes to either duration or concentration of exposure or both. Thus different measures may produce different results (Walker and Blettner 1985). Ideally, the summary measure selected should be based on a set of defensible assumptions regarding the postulated biological mechanism for the agent or disease association under study (Smith 1987). This procedure is not, however, always possible. Very often, the biological effect of the duration of exposure or the concentration of the agent under study is unknown. In this context, the use of different exposure measures may be useful to suggest a mechanism by which exposure exerts its effect.
It is recommended that, in the absence of proved models for assessing exposure, a variety of summary worklife exposure measures be used to estimate risk. This approach would facilitate the comparison of findings across studies.
" DISCLAIMER: The ILO does not take responsibility for content presented on this web portal that is presented in any language other than English, which is the language used for the initial production and peer-review of original content. Certain statistics have not been updated since the production of the 4th edition of the Encyclopaedia (1998)."